АНОМАЛИИ МЕЖЖЕЛУДО ЧКОВОЙ ПЕРЕГОРОДКИ 5 страница
Сократительный аппарат мышечных волокон сердца состоит из продольно ориентированных миофибрилл, которые образованы последовательно соединенными саркомерами, являющимися основной структурной и функциональной единицей миокарда (рис. 44). Саркоплазма миофибрилл содержит центрально расположенное ядро, большое число митохондрий, служащих основным источником энергообразования, и саркоплазматический ретикулум, являющийся структурной основой сопряжения возбуждения и сокращения.
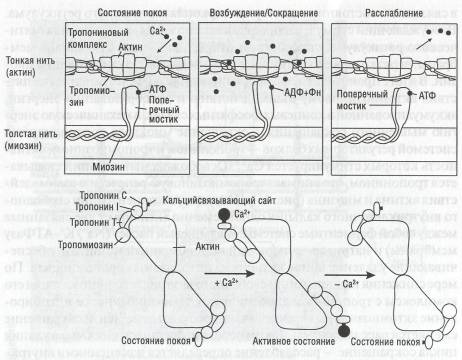
Своей способностью развивать усилие и укорачиваться мышца обязана структуре саркомера, длина которого строго пропорциональна длине мышцы (рис. 45). В саркомере различают диски, актиновые и миозиновые нити, тропомиозин, тропониновые комплексы. Границей между двумя саркомерами является диск Z, от которого отходят тонкие нити актина. Область диска А (внутри саркомера) состоит из толстых нитей миозина. В зоне Н диска А нити актина прерываются, и она представлена только нитями миозина. Часть саркомера между концами миозиновых нитей составляет диск I, который состоит только из тонких нитей актина. Организация сократительных белков в миофиб-риллах служит основой механизма мышечного сокращения.
 330
330
Некоронарогенные болезни сердца
 Рис. 44. Схема структуры мышцы сердца (по F. Netter, 1969, с изменениями)
Рис. 44. Схема структуры мышцы сердца (по F. Netter, 1969, с изменениями)
|
|

| Некоронарогенные болезни сердца |
Рис. 45. Схема сокращения и расслабления мышечного волокна (по F. Netter, 1969, с изменениями)
|
|
|
Миозин способен расщеплять АТФ, следствием чего является выделение энергии для сокращения, а также обратимо связываться с актином в актомиозиновый комплекс, что проявляется сокращением мио-фибрилл. Ферментативные свойства миозина и способность его связываться с актином активируются Са++. В состоянии покоя Са++ находится
332
Некоронарогенные болезни сердца
в связанном состоянии в цистернах саркоплазматического ретикулума. Побуждающий СТИМул, распространяясь по канальцам саркоплазмати-ческого ретикулума, способствует открытию L-типа Са-каналов мем-баны, черезкоторыевклеткупоступаютионыСа++,акгивирующиемио-зин В итоге происходит дефосфорилирование АТФ, устранение препятствия актомиозиновому взаимодействию и преобразованию энергии, аккумулированнойвконцевыхфосфатныхсвязях,вмеханическую энергию мышечного сокращения. Сокращение миокарда контролируется системой регуляторных белков - тропонином и тропомиозином, активность которых стимулируется Са++. Освобождаемый кальций связывается тропонином, что снимает тропомиозиновую репрессию взаимодействия актина и миозина (рис. 46). Повышение концентрации свободного внутриклеточного кальция одновременно активирует две связанные между собой ферментные системы: «кальциевый насос» (Na+/K+-АТФазу мембраны) и натриево-кальциевый ионообменный механизм, обеспечивающие удаление ионов кальция из цитоплазмы кардиомиоцита. По мере снижения концентрации свободного внутриклеточного кальция его комплексы с тропонином диссоциируют, тропомиозиновое ингибиро-вание актомиозиновых взаимосвязей восстанавливается, и сокращение сменяется расслаблением. Таким образом, физиологическая регуляция цикла сокращение - расслабление определяется изменениями внутриклеточной концентрации ионов Са++.
|
|
|
При дилатационной кардиомиопатии найдены мутации генов, кодирующих белки внеклеточного матрикса, что может служить причиной ослабления механической взаимосвязи между последним и кардио-миоцитами, возможным следствием чего является прогрессирующая дилатация сердца. В кардиомиоцитах происходят изменения в экспрессии генов, которые затрагивают контрактильные белки или их регулирующие элементы, а также различные механизмы, обеспечивающие сопряжение процессов возбуждения - сокращения, бета-адренер-гические пути проведения и процессы, приводящие к дефициту энергетических механизмов.
|
|
|
Сила сокращения мышечных волокон зависит от их исходной длины (закон Франка - Старлинга), что является решающим фактором,
333
|
|
| Некоронарогенные болезни сердца |
Рис . 46. Взаимодействие сократительных белков и схема сокращения мышечного волокна (по Е. Браунвальду, 1995, с изменениями)
определяющим функцию сердечной мышцы. При фиксированной исходной длине миокардиальных волокон реализуется инотропный механизм, который является детерминантой качественной характеристики процесса мышечного сокращения. Влияя на интенсивность преобразования химической энергии дефосфорилирования АТФ в механическую энергию сокращающейся мышцы на этапе формирования акто-миозиновых контактов, он регулирует и силу, и скорость сокращений миокардиальных волокон, изменяя их сократимость. В условиях положительных инотропных влияний возникает рост мощности миокарда и выполняемой им работы.
Некоронарогенные болезни сердца
Ударный объем интактного желудочка в конечном счете определяется тремя факторами:
1) длиной мышцы в начале сокращения — преднагрузкой,
2) инотропным состоянием мышцы (сократимостью)— состоянием
связей сила — скорость — длина,
|
|
|
3) напряжением, которое должна развивать мышца во время сокра
щения, то есть постнагрузкой (схема 4).
Схема 4. Взаимодействие различных компонентов, регулирующих сердечную деятельность
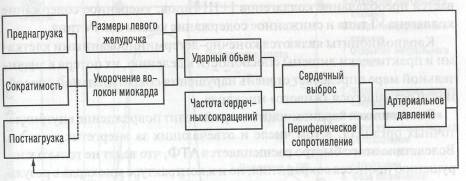
Основным критерием сократительного состояния миокарда служит развиваемое им напряжение — формируемая волокнами сила (во время выброса крови), действие которой направлено на их укорочение, отнесенная к суммарному поперечному сечению миокардиальных волокон. Источником этой силы является энергия, образующаяся в миокарде вследствие окисления и аккумулируемая главным образом в форме АТФ. Напряжение миокарда левого желудочка способствует сокращению стенки желудочка, уменьшению его полости и изгнанию из него крови. Если функция левого желудочка начинает нарушаться, то есть нет резерва преднагрузки, то в поддержании функции сердца значительно возрастает значение постнагрузки левого желудочка.
При дилатационной кардиомиопатии происходит диффузное поражение миокарда с развитием деструкции кардиомиоцитов и формированием заместительного фиброза. Определенная роль в развитии фиб-
335
Некоронарогенные болезни сердца
Некоронарогенные болезни сердца
роза отводится деградации нормального коллагенового матрикса металлопротеиназами, активация которых происходит вследствие активации провоспалительных цитокинов и экспозиции свободных кислородных радикалов (оксидантного стресса). Одновременно происходит аномальный синтез коллагена фибробластами. Вновь образуемые коллагеновые структуры характеризуются извращенным соотношением между типами коллагена и нарушением архитектоники взаиморасположения волокон. При дилатационной кардиомиопатии обнаруживается преобладание коллагенов I+III типов, умеренное содержание коллагена VI типа и сниженное содержание коллагена IV типа.
Кардиомиоциты являются конечно-детерминированными клетками и практически лишены способности к делению, их потеря в значительной мере определяет степень нарушения сократительной способности оставшегося «живого» миокарда.
В оставшихся кардиомиоцитах происходит повреждение внутриклеточных органелл, в том числе и отвечающих за энергетику клетки. Вследствие этого быстро расщепляется АТФ, что ведет не только к нарушению процесса сокращения, но и к контрактуре миокарда в результате нехватки энергии и кальциевой перегрузки. При этом одновременно значительно снижаются показатели как систолы, так и диастолы. Процесс наполнения желудочков характеризуется существенным изменением временных, скоростных и объемных показателей всех фаз диастолы, «вклад» систолы предсердий в наполнение желудочков отсутствует уже на самых ранних этапах декомпенсации. Нарушение систолической функции левого желудочка проявляется в уменьшении максимальной скорости укорочения миокардиальных волокон во время их возбуждения, в снижении скорости нарастания внутрижелудоч-кового давления и достигаемого максимума давления в период напряжения миокарда, в удлинении периода напряжения и укорочении периода изгнания (изменения фазовой структуры систолы) (рис. 47). Увеличение объема остаточной крови приводит к постепенному расширению полости левого желудочка.
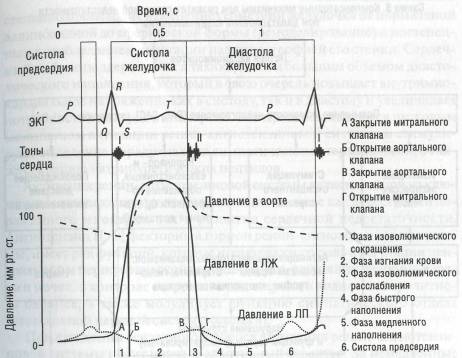
Рис. 47. Схематическое изображение сердечного цикла
При повреждении миокарда и дилатации сердца происходит активация различных компенсаторных механизмов, направленных на нормализацию сердечной деятельности (схема 5).
Благодаря системе нейрогуморальной регуляции сердце в течение определенного периода способно поддерживать насосную функцию при сниженной сократительной способности миокарда с помощью срочных механизмов компенсации гемодинамики, к которым относятся активация механизма Франка — Старлинга и повышение хроно- и инотропной активации миокарда.
Долговременные компенсаторные механизмы включают развитие гипертрофии оставшихся жизнеспособных кардиомиоцитов и измене-
Некоронарогенные болезни сердца
Схема 5. Компенсаторные механизмы при развитии сердечной недостаточности при дилатационной кардиомиопатии
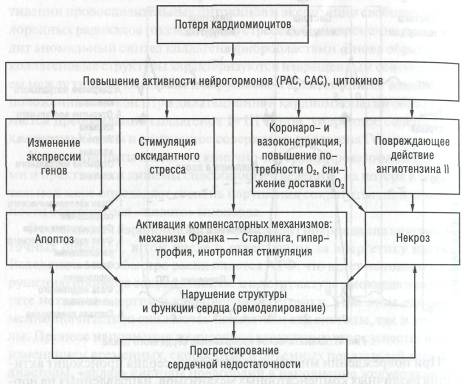
ние геометрии камер сердца, что составляет суть ремоделирования левого желудочка. Дилатация желудочка служит ранним компенсаторным ответом на уменьшение сократимости и кинетики стенок, способствуя сохранению полноценного ударного объема за счет увеличения конечного диастолического объема.
Гипертрофия миокарда у больных с дилатационной кардиомиопа-тией не достигает адекватной степени и не отвечает нуждам дилатиро-ванного сердца, поскольку сократительная активность гипертрофированного миокарда на единицу массы ниже, чем в здоровом сердце. По-
Некоронарогенные болезни сердца
степенно происходит изменение геометрии желудочка от нормальной эллипсоидной до сферической формы (ремоделирование) и постепенным преобладанием дилатации над гипертрофией его стенки. Сердечный выброс поддерживается тахикардией и большим объемом диастолического наполнения, который в свою очередь повышает внутримио-кардиальное напряжение как в систолу, так и в диастолу и увеличивает потребность миокарда в кислороде. Эти процессы являются пусковым механизмом активации ренин-ангиотензиновой системы и стимулируют выделение норадреналина симпатическими терминалями, а также секрецию натрийуретических пептидов.
Активация ренин-ангиотензиновой системы является одной из ключевых составляющих процессов, лежащих в основе как перестройки пораженного миокарда, так и развития сердечной недостаточности. Ангиотензин II, эффекторный гормон ренин-ангиотензиновой системы, имеет ряд функций, которые играют важную роль в регуляции центральной и периферической гемодинамики, регуляции функции сердца и почек, в контроле секреции альдостерона и водно-электролитного баланса, а также модулирует функцию симпатического отдела вегетативной нервной системы (схема 6).
При снижении сердечного выброса активация ренин-ангиотензиновой системы носит вначале адаптивный характер. За немедленные реакции (вазоконстрикцию, задержку натрия и воды, инотропную и хронотропную реакции) ответственна циркулирующая система, за хронические реакции (митогенное усиление роста клеток, гипертрофию, апоптоз) ответственна тканевая система, находящаяся непосредственно в миокарде.
Длительная чрезмерная активация ренин-ангиотензиновой системы оказывает повреждающее действие, следствием чего является:
• потенцирование активности других нейрогормональных систем —
симпатоадреналовой, эндотелина, также играющих важную роль в раз
витии апоптоза и некроза, клеточного роста и ремоделирования;
• сосудистые эффекты — коронарная и системная вазоконстрикция,
провоцирующие увеличение нагрузки на сердце и потребность мио
карда в кислороде, что в комбинации с задержкой жидкости ведет к
339
Мнявмвм
Некоронарогенные болезни сердца
Схема 6. Регуляторные зффекты ангиотензина II в условиях сниженного сердечного выброса
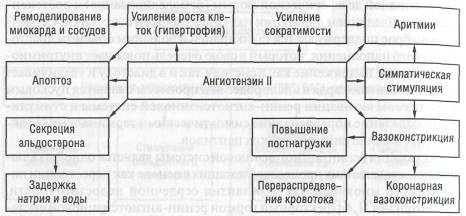
гемодинамической перегрузке и перерастяжению стенки пораженного миокарда и служит пусковым моментом к развитию апоптоза, изменению генной экспрессии и ремоделирования;
• непосредственное токсическое повреждающее действие ангиотензина II на кардиомиоциты, что приводит к их дисфункции и гибели.
Ангиотензин II является самостоятельным триггерным фактором реакций генетического ответа и клеточного роста, что вызывает гипертрофию кардиомиоцитов и гиперплазию фибробластов (развитие фиброза). Вследствие развития фиброза происходит уменьшение плотности капилляров миокарда, приводящее к его ишемизации.
Активация симпатоадреналовой системы у больных с дилатацион-ной кардиомиопатией носит вначале компенсаторный характер, обеспечивая насосную функцию сердца путем повышения частоты сокращений сердца и сократимости миокарда, поддержания артериального давления в условиях сниженного сердечного выброса, вызывая кон-стрикцию артериол, потенцируя веноконстрикцию, обеспечивая венозный возврат и повышение давления наполнения сердца и сердечный выброс посредством механизма Франка — Старлинга. В дальнейшем симпатическая активация играет важную роль в прогрессировании сердечной недостаточности (схема 7).
Некоронарогенные болезни сердца
Схема 7. Роль симпатоадреналовой системы в патогенезе прогрессирования хронической сердечной недостаточности при дилатационной кардиомиопатии
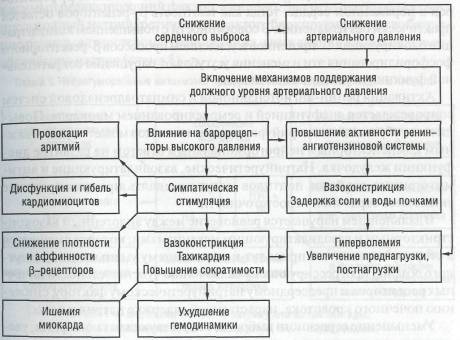
Длительная активация симпатоадреналовой системы оказывает ряд негативных эффектов на сердечно-сосудистую систему, включая повышение потребности в кислороде, депрессию силы сокращения (вследствие увеличения частоты сердечных сокращений), снижение чувствительности β-an-ренорецепторов к катехоламинам, повышение уровня ренина и ангиотензина II, прямое токсическое воздействие на миокард (перегрузка кардиомиоцитов Са++, прогрессирующая гибель клеток миокарда в результате некроза и апоптоза, угнетение функции митохондрий (окислительно-восстановительное напряжение), что опосредуется через β-peцeптopы сердца и цАМф, повышается концентрация катехоламинов в плазме крови.
Описанные изменения подкрепляются нарушениями со стороны β-aдpeнepгичecкиx путей проведения, в значительной мере модулирующих
Некоронарогенные болезни сердца
функцию сердца на рецепторном и клеточном уровнях. В результате происходит значительное уменьшение количества и плотности β1-peцenтo-ров в пораженном сердце, тогда как плотность β2-рецепторов остается практически без изменений. В совокупности с повышением концентрации блокирующих G-протеинов и усилением процессов β-рецепторного фосфорилирования эти изменения усугубляют нарушения сократительной функции пораженного миокарда.
Активация ренин-ангиотензиновой и симпатоадреналовой систем сопровождается дисфункцией и ремоделированием миокарда. Повышенное образование натрийуретических пептидов может служить важным компенсаторным нейрогормональным ответом на развитие дисфункции желудочка. Натрийуретические, вазодилатирующие и анти-митогенные свойства пептидов могут замедлять прогрессирование симптомов сердечной недостаточности.
В дальнейшем нарушается равновесие между эндогенным вазокон-стрикторным и вазодилатирующим механизмами, которое смещается в сторону первых, что приводит к дальнейшему уменьшению минутного объема, прогрессирующему ослаблению чувствительности почечных рецепторов к предсердному натрийуретическому фактору, снижению почечного кровотока, нарастающей задержке натрия и воды.
Уменьшение сердечного выброса, сопутствующая тахикардия, увеличение экстраваскулярного компонента коронарного сопротивления (повышение конечно-диастолического давления в левом желудочке) приводят к перераспределению коронарного кровотока, обеднению кровоснабжения субэндокардиальных областей. Измененная геометрия желудочков приводит ко вторичной функциональной митральной илитрикуспидальной регургитации и дилатации предсердий.
Среди причин, приводящих к прогрессирующей альтерации цито-архитектоники миокарда и сосудов, избыточному накоплению внеклеточного матрикса, несбалансированной гипертрофии миокарда и сосудистой стенки при дилатационной кардиомиопатии и сердечной недостаточности рассматривается активация «immediate early» — генов, неспецифических факторов роста (вазопрессин, ангиотензин II, эндо-
342
Некоронарогенные болезни сердца
телин, альдостерон, катехоламины и др.), нарушение продукции вазо-депрессорных субстанций (простациклин, оксид азота, эндотелийза-висимый релаксирующий фактор, кинины). Значительную роль играет предсердный натрийуретический фактор, блокирующий высвобождение ренина, альдостерона, вазопрессина, снижающий канальцевую ре-абсорбцию натрия (схема 8).
Схема 8. Нейрогуморальные механизмы прогрессирования сердечной недостаточности

ЭТ-1 — эндотелин-1, НУП — натрийуретические пептиды, АII — ангиотензин II, N0 — оксид азота, НА — норадреналин.
Процесс ремоделирования периферических сосудов и сердца у больных с сердечной недостаточностью протекает параллельно и подчинен единым патофизиологическим механизмам. Повреждение миокарда с ухудшением сократительной деятельности желудочков сердца приводит к увеличению сосудистого сопротивления, снижению податливости стенок сосудов и дисфункции эндотелия. Последняя способствует нарастанию дилатации, зависимой от кровотока, и увеличению перегрузки сердца.
На определенном этапе перестройка гемодинамики начинает отрицательно сказываться на работе сердца вследствие существования обратной зависимости между системным сосудистым сопротивлением и ударным объемом. Создается порочный круг: снижение сердечного
 343
343
Некоронарогенные болезни сердца
Дата добавления: 2019-03-09; просмотров: 169; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!