Загрязнение осадков. Явление соосаждения, его роль и причины. Адсорбция. Окклюзия. Правила Панета-Фаянса-Гана. Изоморфное соосаждение. Правило Хлопина. На конкретных примерах.
В гравиметрическом определении часто возникают ошибки, связанные с загрязнением осадков. Самый распространенный вариант загрязнения осадков – соосаждение – процесс перехода осадка в раствор или раствора в осадок или процесс распределения микрокомпонента между раствором (жидкая фаза) и осадком (твердая фаза) без образования собственной твердой фазы.
Роль соосаждения: отрицательная: соосаждение – один из источников погрешностей в гравиметрии; положительная: соосаждение используется для концентрирования микрокомпонентов (соосаждение на коллекторе). Соосаждение на коллекторе (сульфиды, гидроксиды металлов) позволяет отделить микрокомпонент от основы. Например, при анализах воды важное значение имеет определение содержания в ней свинца. Между тем концентрация РЬ2+-ионов в воде настолько мала, что величина произведения растворимости даже наименее растворимого из соединений свинца — PbS не может быть достигнута.Чтобы вызвать осаждение Pb2+, нужно во много раз увеличить концентрацию его в растворе. Гораздо быстрее и проще провести концентрирование микрокомпонента путем соосаждения его с каким-либо подходящим коллектором. В данном случае таким коллектором служит осадок СаСОз, образующийся при прибавлении к исследуемой воде раствора Na2CO3. Вместе с CaCO3 соосаждаются практически все присутствующие в воде РЬ2+-ионы. Если отделить образовавшийся небольшой осадок и затем растворить его в возможно малом количестве HCl или CH3COOH, то получится раствор, в котором концентрация РЬ2+-ионов на несколько порядков выше, чем в исходной воде. Определение Pb2+ в этом растворе уже не представляет затруднений.
|
|
|
Различают следующие виды соосаждения: 1) Адсорбция; 2) Окклюзия; 3) Изоморфное соосаждение.
1) Адсорбция - соосаждение примесей на поверхности уже сформированного осадка. Причина адсорбции – неравноценность электростатического поля ионов внутри кристаллической решетки и на поверхности, т.е. ионы или молекулы, расположенные на поверхности твердой фазы, находятся в иных условиях, чем частицы, лежащие внутри нее. Адсорбция характеризуется ярко выраженной избирательностью. Преимущественно из раствора адсорбируются те ионы, которые входят в кристаллическую структуру осадка. В качестве противоионов адсорбируются примеси.
Адсорбция противоионов подчиняется правилам Панета-Фаянса-Гана:
а) при одинаковых концентрациях преимущественно адсорбируются многозарядные ионы;
б) при одинаковых зарядах – ион, присутствующий в большей концентрации;
в) при одинаковых концентрациях и зарядах ион, который с ионом решетки образует менее растворимое, менее диссоциирующее соединение или вообще соединение со значительным межионным взаимодействием;
|
|
|
г) в кислой среде соосаждение катионов уменьшается вследствие конкурентной адсорбции H3O+.
Правило в общем виде: Катионы, адсорбированные поверхностью тем больше, чем меньше растворимость соединения, образующегося из этого катиона и аниона осадка. Количество адсорбированной примеси зависит от величины поверхности осадка, концентрации адсорбирующейся примеси и температуры. С увеличением поверхности и концентрации примеси адсорбция возрастает, с повышением температуры адсорбция уменьшается. В растворе может наблюдаться равновесие межу адсорбцией и десорбцией (перенос вещества с поверхности раздела фаз в объем фазы). Пример адсорбции 1: раствор BaCl2приливают к раствору H2SO4. После осаждения систем имеет состав:BaSO4, Cl-, Ba2+, H3O+. В этих условиях осадок BaSO4 адсорбирует первоначально одноименный ион Ba2+, а в качестве противоиона Cl-, т.е. загрязняется BaCl2. Пример адсорбции 2: раствор BaCl2 прибавляют к раствору, содержащему H2SO4 и NH4NO3. После осаждения систем имеет состав:BaSO4, Cl-, Ba2+, NO3-, NH4+, H3O+. В этих условиях осадок BaSO4 адсорбирует первоначально одноименный ион Ba2+, а в качестве противоиона NO3-, т.е. загрязняется Ba(NO3)2, т.к. нитрат бария менее растворим, чем хлорид бария.
|
|
|
2) Окклюзия – загрязнение осадка в результате захвата примесей внутрь растущих кристаллов осадка основного компонента. Причина окклюзии – адсорбция примесей в процессе формирования осадка.
Виды окклюзии: 1) Адсорбционная. Возникает при быстром росте кристаллов, когда ионы на поверхности обрастают кристаллизационным веществом. Протекает вследствие адсорбции примесей по микротрещинам кристаллической структуры. 2) Механическая – случайный захват частиц маточного раствора внутрь осадка. Характерна при выделении аморфных осадков. Окклюдированные примеси равномерно распределяются внутри, но не принимают участия в построении кристаллической решетки. Окклюзия подчиняется тем же правилам, что и адсорбция вследствие того, что в ее основе лежат адсорбционные явления. Понизить окклюзию можно путем замедления процесса выделения твердой фазы, т.е. осаждение проводят при малом пересыщении (осадитель прибавляют по каплям). Пример окклюзии: раствор BaCl2 вливают в раствор H2SO4. В процессе осаждения система имеет состав:BaSO4, Cl-, SO42-, H3O+. В этих условиях осадок BaSO4 окклюдирует прежде всего собственный ион SO42-, а в качестве противоионаH3O+, т.е. загрязняется H2SO4 вследствие адсорбционной окклюзии.
|
|
|
3) Изоморфное соосаждение – образование смешанных кристаллов (образование твердого раствора). Характерно для изоморфно-кристаллизующихся веществ, которые могут образовывать смешанные кристаллы. Наблюдается тогда, когда вещества сходны по химическим свойствам или ионы имеют одинаковые координационные числа и радиусы (условием изоморфного соосаждения является равенство зарядов и близость ионных радиусов макро- и микрокомпонентов).
Подчиняется правилу Хлопина: если оба вещества являются изоморфными и концентрация одного из них мала, то распределение их микрокомпонентов между кристаллической фазой и раствором при постоянной температуре характеризуется постоянной величиной и не зависит от количественного соотношения фаз:  ,
,  . Примеры веществ, для которых характерно изоморфное соосаждение:ZnCO3, MgCO3, FeCO3, MnCO3, CdCO3, CaCO3; BaSO4, PbSO4, SrSO4, RaSO4; MgNH4PO4, MgKPO4; MnNH4PO4, ZnNH4PO4; ZnHg(SCN)4, CoHg(SCN)4, NiHg(SCN)4.
. Примеры веществ, для которых характерно изоморфное соосаждение:ZnCO3, MgCO3, FeCO3, MnCO3, CdCO3, CaCO3; BaSO4, PbSO4, SrSO4, RaSO4; MgNH4PO4, MgKPO4; MnNH4PO4, ZnNH4PO4; ZnHg(SCN)4, CoHg(SCN)4, NiHg(SCN)4.
Другие виды загрязнения: 1) Совместное осаждение – выделение в твердую фазу нескольких веществ, для которых в условиях осаждения достигнуты величины их KST. 2) Последующее осаждение – выделение примеси на поверхности уже сформировавшегося осадка.
Методы отгонки. Вещества, определяемые методом отгонки. Прямые и косвенные методы отгонки. Поглотители для прямого метода отгонки. Методы определения карбонатов в карбонатных породах.
Метод отгонки – метод гравиметрического анализа, основанный на выделении определяемого компонента в виде летучего соединения с последующим определением массы отогнанного вещества или массы остатка.
Вещества, определяемые методом отгонки:
1) карбонаты и углекислый газ в карбонатных породах;
2) кристаллизационная вода в гидратах солей;
3) бор в виде летучих эфиров борной кислоты, например, B(OCH3)3; 2) мышьяк в виде AsCl3; 3) германий в виде GeCl4;
4) железо в виде FeCl3;
5) ртуть в виде Hg и т.д.
Метод отгонки также применяют при определении ванадия, вольфрама, молибдена, олова, сурьмы, иода, фтора, осмия, серы, селена, теллура, кремния и других элементов. Отгонка SiF4 – отделение кремниевой кислоты от других примесей (соединений титана, железа, щелочных и щелочноземельных металлов и др.).
Косвенные методы отгонки – методы, при которых определяется масса остатка. Определяемое вещество отгоняют из точной навески анализируемого образца. После окончания отгонки образец снова взвешивают. Массу определяемого вещества находят по разности масс образца до и после отгона. В пищевой промышленности таким образцом определяют массовую долю влаги в различных видах сырья и готовых изделиях. Например, в хлебопекарной промышленности анализируют на этот показатель муку, практически все дополнительное сырье (сухое коровье молоко, сгущенное молоко с сахаром, сливочное масло и т.д.), а также готовые хлебобулочные изделия. Пример: определение кристаллизационной воды в кристаллогидрате хлорида бария:BaCl2*2H2O=BaCl2+2H2O↑.
Прямые методы отгонки – методы отгонки, при котором определяемое вещество отгоняют из смеси и затем определяют массу отогнанного вещества или определяемый летучий компонент поглощают специфическим поглотителем и по увеличению массы поглотителя вычисляют содержание определяемого компонента. Пример: определение карбонатови CO2 в карбонатных породах CaCO3+H+=Ca2++CO2↑+H2O, CO2+2NaOH=Na2CO3+H2O (поглотитель в данном случае – натронная известь CaO+NaOH).Процентное содержание карбоната вычисляют по формуле: ω =  , где mосадка - масса высушенного или прокаленного осадка, г;F–гравиметрический фактор, a – навеска анализируемого вещества.
, где mосадка - масса высушенного или прокаленного осадка, г;F–гравиметрический фактор, a – навеска анализируемого вещества.
Вещества-поглотители: натронная известь CaO+NaOH (поглощает CO2), хлорид кальция CaCl2 и оксид фосфора (V)P2O5 (поглощают воду).
15. Титриметрический кислотно-основный метод определения HCl в техническом продукте: возможные титранты, стандартизация рабочего раствора, первичные и вторичные стандарты, кривая титрования HCl, возможные индикаторы, индикаторные погрешности.
Титриметрический метод анализа – это метод, основанный на измерении объема раствора реактива известной концентрации, затраченного на реакцию с определяемым компонентом. В титриметрическом методе анализа количество химических веществ определяется путем точного измерения объемов растворов двух веществ, вступающих между собой в определенную реакцию. Раствор реактива (титрованный раствор или стандартный раствор) добавляют к анализируемому раствору до тех пор, пока количество прибавленного реактива не станет эквивалентным количеству реагирующего с ним определяемого компонента.
Возможные титранты: гидроксид натрия NaOH, стандартный, 0,100 моль-экв/л раствор.
Стандартизация рабочего раствора, первичные и вторичные стандарты. Поскольку в титриметрическом анализе содержание определяемого вещества рассчитывают по объему титрованного раствора, израсходованного на титрование, от тщательности приготовления последнего зависит успех анализа. Концентрация титрованного раствора (его титр) должна быть определена очень точно.
В титриметрическом анализе пользуются титрованными растворами: первичными и вторичными стандартами.
В тех случаях, когда вещество, применяемое для приготовления раствора строго соответствует химической формуле и является химически чистым, навеску этого вещества можно рассчитать в соответствии с заданным титром раствора и отвесить его на аналитических весах с точностью до 0,0002 г.
Навеску для приготовления раствора рассчитывают по формуле: 
Взвешенное вещество помещают в откалиброванную мерную колбу, растворяют в воде или другом подходящем растворителе, доводят объем раствора до метки и тщательно перемешивают. Приготовление растворов известной концентрации требует соблюдения очень точных приемов работы и большого опыта.
В тех случаях, когда вещество не может быть получено в чистом виде или когда оно отличается неустойчивостью (легко теряет кристаллизационную воду, подвергается действию углекислого газа или воздуха, вступает во взаимодействие с примесями, содержащимися в воде и т.д.), из него готовят растворы приблизительной концентрации (беря навеску на технических весах с точностью до 0,01 г). Затем их стандартизуют, т.е. устанавливают истинную концентрацию, либо по так называемым исходным веществам (первичным стандартам), либо по рабочим растворам с уже точно определенной концентрацией (вторичным стандартам). Проверяют концентрацию приготовленного раствора и выводят коэффициент поправки К:  .
.
Концентрацию рабочего раствора NaOH устанавливают либо по вторичному стандарту – титрованному рабочему раствору HCl, либо по первичным стандартам (щавелевой, янтарной, бензойной кислотам, гидрофталату калия и др.).
Определение К раствора NaOH по раствору HCl методом пипетирования. В коническую колбу помещают 15,0-205,0 мл титрованного рабочего раствора хлороводородной кислоты 0,100 молярной концентрации эквивалентов, добавляют 2-3 капли метилового оранжевого и титруют рабочим раствором NaOH до перехода окраски из красной в желтую. Титрование повторяют 3 раза, усредняют три значения объема титранта, отличные не больше чем на 0,1 мл, и рассчитывают КNaOH: 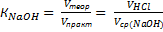 .
.
Определение К раствора NaOH по щавелевой кислоте методом пипетирования. КонцетрациюNaOH устанавливают по перекристаллизованной щавелевой кислоте. Препарат необходимо перекристаллизовывать, т.к. он содержит две молекулы воды, которые частично выветриваются при хранении. Кроме того, пользуются свежеприготовленным раствором щавелевой кислоты, так как концентрация последнего уменьшается с течением времени. Между щавелевой кислотой и гидроксидом натрия протекает следующая реакция:H2C2O4·2H2O+2NaOH=Na2C2O4+4H2O, fэкв(H2C2O4·2H2O)=1/2; Mэкв(H2C2O4·2H2O)=fэкв(H2C2O4·2H2O) ·Mэкв(H2C2O4·2H2O) = ½·126,07 = 63,065 г/моль-экв. Устанавливают коэффициент поправки методом пипетирования. С этой целью в мерной колбе емкостью 200,0-250,0 мл готовят по точной навеске 0,100 моль-экв/л раствор щавелевой кислоты.
Щавелевая кислота слабая двухосновная, но т.к. KIA:KIIA< 104, то нельзя оттитровать каждый протон в отдельности. При титровании используют слабый индикатор с pT>4, например фенолфталеин. Титруют до исчезновения малиновой окраски. Если рабочий раствор NaOH поглотил CO2 и содержит карбонат-ионы, титрование сопровождается переходом Na2CO3и NaHCO3, последний необходимо разрушить. Для этого после обесцвечивания титруемый раствор нагревают, гидрокарбонат натрия при этом гидролизуется с выделением NaOH, окраска фенолфталеина восстанавливается. Раствор дотитровывают до полного обесцвечивания. Процесс нагревания и последующего титрования продолжают несколько раз до полного исчезновения окраски при нагревании титруемого раствора. Навеску щавелевой кислоты рассчитывают для приготовления объема 200,0-250,0 мл раствора 0,100 моль-экв/л:
m(H2C2O4·2H2O) = C(HCl) ·VHC·Mэкв(H2C2O4·2H2O) ·10-3. На аналитических весах взвешивают рассчитанную массу навески гидрата щавелевой кислоты. Если навеска, взятая на аналитических весах, несколько отличается от теоретически рассчитанной, вводят коэффициент пересчета:  . Взятую навеску пересыпают в мерную колбу на 200,0-250,0 мл через стеклянную воронку. Оставшиеся частички щавелевой кислоты смывают с воронки дистиллированной водой. Воронку убирают, в колбу наливают воды примерно до 2/3 от общего объема колбы. Растворяют щавелевую кислоту при перемешивании. Доводят раствор в колбе до метки дистиллированной водой.
. Взятую навеску пересыпают в мерную колбу на 200,0-250,0 мл через стеклянную воронку. Оставшиеся частички щавелевой кислоты смывают с воронки дистиллированной водой. Воронку убирают, в колбу наливают воды примерно до 2/3 от общего объема колбы. Растворяют щавелевую кислоту при перемешивании. Доводят раствор в колбе до метки дистиллированной водой.
В коническую колбу пипеткой отбирают аликвоту 15,0-25,0 мл рабочего раствора NaOH, добавляют 2-3 капли фенолфталеина, титруют 0,100 моль-экв/л раствором щавелевой кислоты до обесцвечивания. Колбу с титруемым раствором нагреваю на плитке, но не кипятят, в случае появления малинового окрашивания горячий раствор дотитровывают до исчезновения окраски. Учитывают общий объем щавелевой кислоты, израсходованной на титрование.
Определение повторяют несколько раз до получения минимум трех значений объема титранта, отличающихся не более чем на 0,1 мл.
Усредняют значение объема титранта и рассчитывают коэффициент поправки: КNaOH=  .
.
Расчет кривой титрования:
Указание веществ, существующих в системе в тот или иной момент титрования:
1.До начала титрования – V(HCl)=100% V(NaOH)=0%pH=1,0
2.До ТЭ:
V(HCl)=50% V(NaOH)=50%pH=1,48
V(HCl)=1% V(NaOH)=99% pH=3,3
V(HCl)=0,1% V(NaOH)=99,9% pH=4,3
3.ВТЭ –V(HCl)=0% V(NaOH)=100% pH=7,0
4.После ТЭ:
V(HCl)=0,1% V(NaOH)=100,1% pH=9,7
V(HCl)=1% V(NaOH)=101% pH=10,7
V(HCl)=10% V(NaOH)=110% pH=11,7
Представление формул для расчета:
1)До начала титрования: pH= - lgC0
2)До ТЭ: pH=  =
= 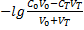 =
= 
3)В ТЭ: pH=pOH=  =7,0
=7,0
4)После ТЭ: pH=  =
=  =
=  при условии С0=СT
при условии С0=СT
Кривая титрования:

Возможные индикаторы:
| Индикатор | Структурная формула | рТ | Интервал перехода рН и окраска индикатора |
| Метиловый оранжевый | 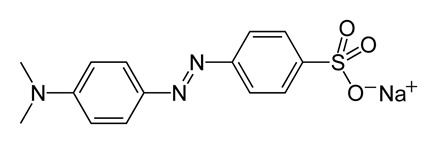
| 4,0 | 3,1–4,4 Красная – оранжево-желтая |
| Бромтимоловый синий | 
| 7,0 | 6,0–7,6 Желтая – синяя |
| Фенолфталеин | 
| 9,0 | 8,2–10 Бесцветная – фиолетовая |
Индикаторные погрешности: Индикаторные погрешности – это погрешности обусловленные несоответствием между показателем индикатора и рН в точке эквивалентности.
Положительная ошибка – избыток титранта.
Отрицательная ошибка – избыток титруемого вещества.
Индикаторные погрешности:
1. Водородная (гидроксониевая)
 (без учета разбавления)
(без учета разбавления)
 (с разбавлением)
(с разбавлением)
2. Гидроксильная ошибка
 (без учета разбавления)
(без учета разбавления)
 (с разбавлением)
(с разбавлением)
3. Кислотная или НА-ошибка

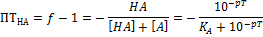
4. Основная или В-ошибка

Условия титрования HCl: исследуемы раствор получают в мерную колбу, доводят до метки водой, тщательно перемешивают. Пипеткой отбирают аликвоту задачи в коническую колбу для титрования, добавляют 2-3 капли метилового оранжевого и тируют рабочим раствором NaOH до перехода окраски из красной в желтую. Титрование потворяют несколько раз, усредняют 3 приемлемых результата. Содержание HCl рассчитывают по формуле:m(HCl)=C(NaOH)*Vср(NaOH)*K(NaOH)*Mэкв(HCl)*P*10-3.
16. Титриметрический кислотно-основный метод определения солей аммония в техническом продукте: возможные титранты, стандартизация рабочего раствора, первичные и вторичные стандарты, способы титрования, условия титрования слабых протолитов, возможные индикаторы.
Дата добавления: 2016-01-05; просмотров: 78; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!