Различают химические (экзогенные и эндогенные) и физические бластомогенные факторы.
Экзогенные химические канцерогены:
· Полициклические углеводороды (бензпирен, дибензантрацен, метилхолантрен) – обладают электрофильными участками молекулы, где находятся атомы с дефицитом электронов, взаим-ют с ДНК. (профессиональный рак в угольной промышл-ти, нефтехимии, резинотехнической промышленности), также явл-ся основными канцерогенами табачного дыма.
· Табак – полицикл. углеводороды образуются в процессе курения; сигаретный дым содержит смесь различных канцерогенов (полицикл. угл, радиоактивный полоний, мышьяк). В случае резорбтивного действия полицикл. углеводородов, печень превращает их в мутагенные эпоксидные соединения, взаим-щие с гуанином ДНК.
· Ароматические амины (нафтиламин) – гистологические красители – явл-ся проканцерогенами и канцерогенами. Глюкоронидаза мочи превращает синтезированный в печени нетоксичный глюкуронид этого канцерогена обратно в активный. Часто поражаются мочевой пузырь, реже – печень, еще реже – почки.
· Нитрозосоединения – в организме эти проканцерогены формируют электрофильный ион – алкил-диазоний. Токсичны для пищевода, печени, в меньшей степени – для других органов. Канцерогенные нитрозамины и нитрозомочевинопроизводные могут возникать в ЖКТ при употреблении нитрит-содержащих продуктов (копчения, консервация)
· Цитостатики – т.к. они облигатно взаим-ют с ДНК, обладают бластомогенной акт-тью. Могут приводить к экспрессии протоонкогенов.
· Тяжелые металлы – разные механизмы, практически все способны образовывать комплексные соединения с ДНК и нуклеопротеидами. Железо повреждает ДНК свободными радикалами. Хроническое мышьяковое отравление приводит к развитию ангиосарком печени. Никелевый рак развивается у работников цветной металлургии.
· Асбест – канцероген и коканцероген. Может изначально сорбировать в процессе обработки и транспорта канцерогены. Приводит к раку легких и образ-нию мезотелиом плевры и брюшины. Механизм действия связывают с хроническим повреждением тканей игольчатыми кристаллами, выделением ростовых факторов макрофагами, разобщением тканевых слоев.
· Терпены (сахарин и цикламаты) – коканцерогены.
· Афлатоксины – антибиотики грибка аспергилла. Обезвреживая проканцерогенный токсин, печень окисляет его в промежуточный продукт – эпоксид, способный взаим-ть с гуанином ДНК и вызывать мутации. Может вызвать некроз гепатоцитов, а при этом стимул-ся компенсаторное выделение факторов роста.
· Хлорвинил (газ) – исп-ют при производстве пластмасс. С одной стороны – причиной канцерогенеза явл-ся разобщение тканей, вызывающее хронический сигнал для фибробластов и снимающее ингибирование роста. С другой стороны – если пластмасса не инертна и высвобождает в организме свободные радикалы.
· Бензол и его гомологи – канцерогены, обладающие лейкозогенной активностью. При летальной мутации они способствуют развитию панмиелофтиза и гемоцитопений.выжившие мутантные клоны несут аномалии генома, некоторые могут выключить гены-супрессоры опухолевого роста. Действие продуктов распада клеток костного мозга и крови способствует притоку ростовых факторов.
Модели: 1) Ямагива и Ишикава – повторные многократные аппликации дёгти или гудрона на кожу уха кролика вызывали сначали воспаление и гиперплазию в виде гиперкератоза, затем – доброкачест. паппилома, затем- плоскоклеточная карцинома кожи.
Оппенгейм – имплантированные под кожу пластмассовые плёнки вызывали неоплазму.
Эндогенный химический канцерогенез – представления о нем впервые были сформулированы русским онкологом Шабадом; он предполагал, что бластомогенные вещества могут образовываться и накапливаться в самом организме; обратил внимание на сходство некоторых экзогенных химических канцерогенов и естественных метаболитов. Так, метилхолантрен м.б получен из дезоксихолевой к-ты желчи. В кишечнике при гниении белков форм-ся аромат-е амины и индолы, особенно при каловой интоксикации и т.д. Шабад показал, что бензоловые эстракты из печени и пораженных органов умерших от рака более канцерогенны для экспериментальных животных, чем сам бензол; желчь оказалась канцрогенной для мышей, при этом в ней был обнаружен метилхолантрен (Давыдовский). Известны канцерогенные метаболиты тирозина и фенилаланина (избыток фенилаланина у беременных с фенилкетонурией может приводить к трансплацентарному канцерогенезу, не затрагивая мать).
Дизгормональные онкогенез – эндокринные и цитокиновые нарушения могут стать причиной возникновения опухоли. – гормонзависимые/гормончуствительные/гормонпродуцирующие опухоли. Например, рак простаты формируется при наличии дигидротестостерона (деривата андрогенов), что явл-ся тропным и митогенным фактором для функций и роста простаты. Модель Лакассаня – модель рака молочной железы у самцов мышей, индуцированная эстрогенным препаратом фолликулином.
Физические бластомогенные ф-ры можно разделить на механические (модели имплантации инородных тел и при радражении полых органов ЖКТ – модель аденокарцины желчного пузыря у морских свинок по Лэйчу-Буйорду, модель папилломы желудка у крыс по Паппенгейму-Лэримору) и лучевые.
· Инфракрасные лучи – низко мутагенны. Описаны случаи «теплового рака». При регенерации получаемых ожогов происходит выделение ростовых факторов, кот-е способствуют пролиферации неопластических клеток.
· Уфо – сильнее всего ДНК поглощаются УФО с длиной 290-320 нм. УФ выбивает электроны из атомов ДНК, делая молекулы оснований высоко реакционноспособными => может происходить сшивка пиримидинов, вызывающая мутиции. Предраковые явления – гиперкератоз и дисплазия (модель Вадовой ультрафиолетового рака кожи на крысах путем 850-часовой инсоляции в горах Сухуми; модели Вайля и Абрикосова, Роффо – при облучении животных светом ртутной лампы)
· Рентгеновкие лучи – у самого Рентгена был лучевой дерматит. Закон Брегонье и Трибодо для мутагенов: ткани с большей пролиферативной активностью более лучепоражаемы: лимфоидные органы -> гонады, кожа, слизистые, почки, печень, мезенхимальные органы, мышечная, нервная, хрящевая и костная ткани. Чувствительность тканей плода и эмбриона в обратном порядке почти что.
Первая – модель Роло-Лапуэнт, Клюне и Мари - саркома у крыс.
Многократные облучения вызывают образование мутантных клонов клеток, а однократное сильное вызывает только гибель.
ð Лейкозы, рак щитовидной железы
· Радиоактивные лучи - первая модель – по Дилсу-Бэтену 1926, карцинома у мышей при действии бромида радия. Хиросима, Нагасаки – лейкозы. Чернобыль – солидные раки (рак щитовидной железы). Известно, что определнные радионуклиды тропны к определенным органам
По Гофману – ионизирующее излучение может само или через оксиданты, генерируемые в ядре клетки, вызывать мутации, при этом вероятность мутаций дозозависима. Мутации, индуцируемые радиацией, могут быть летальными для клеток, в случае выживания их – образуются соматические мутантные клоны с измененными свойствами. Если радиация способствует экспрессии протоонкогена и депрессии антионкогенов – она может быть канцерогенной.
79. Вирусная и вирусно-генетическая этиологии опухолего роста. Механизмы действия вирусов на клетку. Значение обратной транскриптазы в вирусном канцероногенезе.
Вирусно-генетическая теория возникновения опухолей − 1945 г. Зильбер
Принцип пули и мишени: если вирус заканчивает все стадии репликации в клетке, то он убивает её → онкогенные вирусы не завершают последнюю стадию репликации и интегрируются в геном клетки.
Вирусы:
1. РНК- вирусы спиралевидной формы, которые размножаются в цитоплазме (вирусы лейкоза мышей и кур, саркомы Рауса).
2. РНК- вирусы полиэдральной формы.
Вирусы всех групп, содержащие РНК, называют онкорнавирусами (онкогенными, содержащими РНК) или ретровирусами (из-за способности передавать информацию в обратном направлении — от РНК к ДНК).
3. ДНК- вирусы полиэдральной формы, они размножаются в ядрах клеток (вирусы папилломы кроликов, полиомы, бородавки человека). Свойствава типичны для группы, их объединяют под названием papova (papilloma, polioma, vacuolisation).
4. Крупные ДНК- вирусы (вирус фибромы Шоупа, вирус Яба).
Примеры:
· Вирус герпеса→ рак шейки матки;
· вирус гепатита В → гепатоцеллюлярная карцинома,
· аденовирус → опухоль эпителия ВДП
· Ретровирус лимфомы чел-ка → Т-лимфоцитарный лейкоз.
Онкорнавирусы в ДНК-форме имеются в хромосомах нормальных клеток. Они не проявляются, благодаря генам-репрессорам. Воздействие канцерогенов →активация.
Онкогены и опухолеродность вирусов формируются при помощи:
1. Изменения структуры протоонкогенов→ их нерегулируемость - при неполном захвате протоонкогенов вирусами. Проонкогены в геноме клетки представлены интроном и экзоном, в вирусных геномах только экзоны (потеря существенной регуляторной части клеточных генов).
2. Супер-инфекция вирусом клеток, ранее зараженных слабым штаммом онкогенного вируса (обмен ген. информации между вир.) → ↑онкогенности.
Для встраивания в геном клетки генома онкорнавирусов имеет значение "обратная передача генетической информации". Гершензон (1960) и Темин (1964) показали, что передача наследственной информации возможна не только от ДНК на РНК, но и обратно. Был найден фермент, который по РНК, как по матрице, синтезирует комплементарную ДНК. Он назван обратной транскриптазой (РНК-зависимой ДНК-полимеразой). В РНК- содержащих опухолеродных вирусах обнаружена вирусная обратная транскриптаза, а в геномах клеток — ДНК-копии этих вирусов. Профилактика и лечения опухолей, вызываемых онкорнавирусами, путем подавлением обратной транскриптазы.
80. Онкогенная теория опухолевого роста. Понятие о клеточных и вирусных протоонгогенах, промоторах и онкобелках. Значение онкогенов, промоторных последовательностей, онкобелков в развитии опухолей. Физиологическая роль онкогенов. Понятие об антионкогенах. Многошаговый канцерогенез.
Новообразования (неоплазии) – избыточный, дискоординированный, персистентный, аутохтонный рост клеток, характеризующийся необратимостью, а на молекулярном уровне – расстройством функции генов, регулирующих пролиферативные процессы – протоонкогенов и опухолевых супрессоров. Неоплазия развивается многошаговым путем, на основе ступенчатых генетических изменений моноклона клеток.
Активные формы кислорода, оксид азота и его производные в сочетании с инфекционными патогенными факторами, бактериями и вирусами, являются ключевыми факторами канцерогенеза.
Принципиальное различие между нормальной и опухолевой клеткой заключается в том, что переход от покоя к делению (G0=>G1) первой из них инициируется внешними стимулами, тогда как второй – внутренними стимулами, что делает процесс ее деления автономным. В основе опухолевой трансформации клетки лежат активация онкогенов и повреждение генов-супрессоров.
В опухолевой трансформации клеток, возникающей под влиянием различных индукторов канцерогенеза, принципиально участвуют следующие категории генов:
· Онкогены - стимуляторы функций.
· Гены роста и пролиферации клеток (Myc, Ras, Los, ABL и другие).
· Антионкогены (потеря функции).
· Гены, отвечающие за программированную смерть клетки (апоптоз):
– отменяющие программированную смерть: Bcl-2 (стимуляция функций);
– гены смерти клеток – р53 (потеря функции).
Обязательным механизмом развития неоплазий служит неконтролируемая избыточная экспрессия онкогенов в патологических клонах клеток.
Многие признаки морфологического, иммунологического и биохимического атипизма при неоплазии напоминают черты малодифференцированных зародышевых клеток. Атипизм является результатом действия онкобелков – продуктов онкогенов.
При развитии любого конкретного случая неоплазии необходим многошаговый онкогенез. (Понятие ввел Кнудсон в 1981). Пример: для формирования злокачественной опухоли ретинобластомы необходимы 2 мутационных события, которые могут произойти в разное время и под влиянием различных факторов:
1) Предохраняющий от онкогенеза индуктор апоптоза мутантных клеток, антионкоген Rb1 существует в гомологичных хромосомах в 2 копиях. Одна утрачивается до рождения и при зачатии мутация передается всем клеткам.
2) Другая выпадает в одной из этих клеток при соматической мутации.
Протоонкогены. При повреждении различных органов и тканей, вне зависимости от причин этого повреждения, происходит очень быстрая неспецифическая активация ряда генов, которые были объединены под названием «немедленные гены предранней реакции» (НГПР). Считается, что НГПР способны активировать клеточную пролиферацию/ гибель клеток, в зависимости от количества ростовых сигналов. Ген c - myc является регулятором клеточного размножения, а при его сверхактивации наступает озлокачествление клетки, в связи с чем он был впервые описан, как протоонкоген. Ген кодирует ядерный фосфопротеин, способствующий переходу клетки из G1 в S-фазу митотического цикла. Трансформация протоонкогенов в онкогены приводит к их экспрессии и синтезу онкобелков.
Антионкогены или «супрессоры опухолей». Ген Rb даже в гетерозиготном состоянии предохраняет от развития злокачественной ретинобластомы.
Ген р53 – регулятор генной стабильности, останавливающий митотический цикл в мутировавших клетках в стадии G1. Остановка цикла дает мутантной клетке время для срабатывания репаразных механизмов. Если мутация не репарируется, ген р53 способствует апоптозу клеток.
Вирусные протоонкогены. ДНК-содержащие онкогенные вирусы подразделяются на следующие семейства:
1. Семейство Poxviridae;
2. Семейство Herpes viridae, к которому относится вирус Эпштейн-Барра человеа, вызывающий лимфому Беркитта;
3. Семейство Adenoviridae ;
4. Семейство Papovaviridae, представителями которого являются вирусы папилломы.
Также есть РНК-содержащие онкогенные вирусы.
Аденовирусы располагают генами, кодирующими белки-блокаторы апоптоза, гомологи и индукторы клеточного антиапоптогена. ДНК-содержащие опухолеродные вирусы имеют гены-промоторы, которые способны инициировать транскрипцию генов, следующих в ДНК за ними. При ДНК-зависимом вирусном онкогенезе решающую роль играет встраивание вирусного промотора в геном таким образом, чтобы он соседствовал с клеточными протоонкогенами и мог усиливать его экспрессию. Таким образом, ДНК-содержащие опухолеродные вирусы не располагают вирусными онкогенами (исключение - папиломавирус).
Опухолевые ДНК-вирусы кодируют так называемые онкобелки, которые могут выключать апоптоз, препятствовать действию антионкогена р53.
81. Патогенез злокачественных опухолей. Роль онкобелков в малигнизации клеток. Патогенное действие опухоли на организм. Перенеопластические явления и их механизмы. Антибластомная резистентность организма ("иммунный надзор" и неиммунные факторы резистентности). Понятие "предраковые состояния". Стадии онкогенеза. Понятие доброкачественная/злокачественнная опухоль
Патогенез неоплазии, по Лесли Фоулдсу, всегда предусматривает «опухолевую прогрессию», то есть такую эволюционную дивергенцию опухолевого клона, которая, в целом, увеличивает его разнообразие, выживаемость, резистентность по отношению к действующим в организме факторам отбора (иммунным, лечебным воздействиям и т.д.). Итогом этого будет более или менее быстрое нарастание численности клона и его расселение, включая важнейший отличительный признак злокачественных неоплазм – метастазирование.
В патогенезе неоплазии центральную роль играет аномальная экспрессия онкогенов и синтез кодируемых ими онкобелков, действие которых делает клетки автономными в росте и более или менее атипическими по своим метаболическим, генетическим и культуральным свойствам. Обязательно происходит потеря или выключение опухолевых супрессоров антионкогенов, что сообщает неопластическим клеткам потенциальное бессмертие и делает их более мутабельными, нежели нормальные.
Неопластические клоны, чаще всего, не обладают способностью делиться быстрее, чем нормальные малодифференцированные клетки. Но, так как они неспособны к апоптозу и не нуждаются в управляющем сигнале для начала деления – то без лечения и при недостаточно эффективной иммунной защите постепенно вытесняют нормальные клоны.
Патогенетическая роль онкогенов и онкобелков
Основное звено патогенеза новообразований заключается в действии онкогенов, экспрессируемых в неопластических клетках. В настоящее время обнаружено более 50 онкогенов. Многие онкогены — видоизмененные гомологи нормальных клеточных протоонкогенов.
Для нормальной пролиферации необходимо ростостимулирующее действие факторов роста на клеточные рецепторы. Факторы роста стимулируют размножение, а в ряде случаев блокируют апоптоз клеток-мишеней.
В норме под действием ростовых факторов происходят вовлечение стабильных, находящихся в G0-фазе клеток в митотический цикл или, реже, укорочение цикла лабильных клеток. После воздействия сигналопередающая система ростового фактора, в физиологических условиях, инактивируется.
Ростовые факторы (цитокины паракринного и аутокрииного действия и системно влияющие гормоны), взаимодействуют с мембранными рецепторами клеток-мишеней. После связывания происходит димеризация и стимулируется тирозинкиназная активность этих рецепторов. Тирозинкиназы фосфорилируют и, таким образом, «включают» целый ряд внутриклеточных белков, ответственных за пролиферацию клетки.
В результате происходит активация эстафетного каскада, состоящего из нескольких групп белков: фосфолипаза С, ГТФ-связывающие белки (G-белки), белок Raf-1, белки-циклины и т.д.
Протоонкогены могут:
• кодировать ростовые факторы;
• кодировать рецепторы ростовых факторов;
• кодировать пострецепторные передатчики, управляющие клеточным циклом;
• кодировать блокаторы запрограммированной гибели клеток или нарушать самосборку и контактное ингибирование пролиферации.
Онкогены, активированные или/и мутантные гомологи протоонкогенов, могут, соответственно, нарушать функционирование системы на любом из этих этапов — причём, как правило суть их воздействия состоит в придании росторегулирующему каскаду «хронически-активированного» состояния.
Практически, все онкопротеины нарушают информационную сторону ростовых процессов в трансформируемой клетке.
Продукты некоторых онкогенов являются гомологами естественных факторов роста, в частности тромбоцитарного, фибробластического или колониестимулирующихгемопоэтических факторов.
Большую распространенность при неоплазиях имеют онкобелки-гомологи рецепторов ростовых стимуляторов.
Обширная подгруппа онкопротеинов — это тирозиновые протеинкиназы. Они фосфорилируют по остатку тирозина различные клеточные белки, влияя, в основном — в активирующем направлении, на их функции. Имеется обширный список тирозин-протеинкиназных онкобелков, действующих на мишени внутри клетки.
Важная разновидность онкопротеинов представляет собой ГТФ-связывающие белки, аналоги нормальных клеточных адаптеров, сопрягающих поверхностные рецепторы с внутриклеточными эффекторами.
Наконец, многие протоонкогены имеют продукты, действующие, исключительно, внутри клеточных ядер. Они кодируют ключевые белки-активаторы транскрипции (то есть, внутриядерные посредники действия факторов компетентности ростовых цитокинов, включающих экспрессию ядерных ферментов и рецепторов, обеспечивающих ответ на факторы прогрессии). Влияние подобного онкопротеина заменяет действие фактора компетентности, то есть такого цитокина, как, например, тромбоцитарный фактор роста, так как онкобелок «готовит» ядро к ответу на пролиферативный стимул. Действие ядерных транскрипционных факторов пермиссивно, по отношению к стимуляторам синтеза ДНК.
Какие именно мутации приводят к гиперэкспрессии и/или аномальной экспрессии протоонкогенов?
Активация протоонкогенов может наступить в результате мутации в структуре самого протоонкогена. Это могут быть точковые мутации, приводящие к появлению нонсенс- или миссенс-кодонов или точковые мутации сдвига рамки.
Транслокация протоонкогена в другую часть генома может вызвать его избыточную экспрессию, так как он выводится из-под ингибирующего действия своего бывшего соседа-супрессора или размещается рядом с сильным промотором (энхансером). При этом часто образуются химерные гены и химерные онкопротеииы, являющиеся результатом
слияния транспонированного и местного генов.
Возможна амплификация протоонкогенов, приводящая к их гиперэкспрессии.
Наконец, может возникнуть внесение v -онкогена (вирусного) в клетку на участок, подлежащий промоторному действию клеточных генов (ретровирусный онкогенез), либо наоборот — появление вирусных промоторов вблизи клеточного протоонкогена (онкогенез ДНК-содержащими вирусами).
Роль антионкогенов в патогенезе неоплазии
Немаловажную роль в патогенезе неоплазии играет ограничение и выпадение функций физиологических опухолеподавляющих генов — антионкогенов.
Антионкогены кодируют антионкопротеины, подавляющие пролиферацию. В основном, их продукты — антагонисты онкобелков или индукторы апоптоза. Имеются среди них и адгезивные протеины, обеспечивающие восстановление взаимоотношений пролиферирующих клеток с соседями и межклеточным матриксом, включая контактное ингибирование.
Антионкогены могут утрачиваться в результате делеции генов и более крупных участков хромосомного материала или становиться дефектными, в результате точковых нонсенс- и миссенс-мутаций.
Противоопухолевый иммунитет (Антибластомная резистентность организма)
В патогенезе неоплазм огромную роль приобретают взаимоотношения иммунной системы и клеток новообразования. Этими взаимоотношениями определяются скорость роста опухоли, ход её прогрессии и часть паранеопластических явлений в организме больного. Иммунитет основан на функции лимфоидных клеток, в которых действуют транспозоны и обеспечена повышенная способность к соматическому мутированию. Однако, и в основе неопластической трансформации лежит цепь соматических мутаций. Еще Бернет (1958) указывал, что в филогенезе как высокая частота опухолевого роста, так и сложные формы иммунного ответа проявляются у высших позвоночных параллельно и считал, что между этими явлениями имеется общебиологическая связь, допуская даже, что эволюционное развитие иммунной системы представляло собой ответ на формирование способности к неоплазии (гипотеза иммунологического надзора).
Неопластический процесс, действительно, ведёт к экспрессии ряда антигенов, индуцирующих противоопухолевый иммунный ответ.
Но еще до индукции специфического ответа неопластические клетки приводят в действие факторы естественной неспецифической резистентности к опухолям.
Это, прежде всего, деятельность естественных киллеров — NK -клеток. Данные агенты противоопухолевого иммунитета опознают неопластические клетки по сниженной плотности расположения на плазматической мембране последних антигенов ГКГС. Антигены ГКГС собственных клеток, в норме, являются блокирующими лигандами для так называемых KIR-peцепторов естественных киллеров. Снижение количества ГКГС-молекул, свойственное неопластическим клонам разной природы, служит для NK-клеток дерепрессирующим сигналом и активирует её цитотоксичность.
Неспецифические механизмы противоопухолевой защиты (осуществляются NK-клетками) доминируют на начальных стадиях ответа, когда количество трансформированных клеток не превышает 103.
Если количество опухолевых клеток возрастает до 106 и более — на передний план выходят механизмы противоопухолевого иммунитета, детерминированные антиген-специфическим иммунным ответом.
Основой противоопухолевого иммунитета принято считать реакции замедленной гиперчувствительности, опосредованные цитотоксическими Т- лимфоцитами. Т-иммунитет при опухолевом росте не всегда эффективен и подвержен явлению Т-супрессии.
Большое значение в антинеопластическом иммунитете имеет а-интерферон, угнетающий неопластические клетки. Данный интерферон препятствует васкуляризации опухолей и индуцирует их склерозирование. Интерферон-гамма — продукт натуральных киллеров, проникающих в неоплазму, он активирует цитотоксический потенциал самих инфильтрирующих опухоль NK -клеток, а кроме того — нейтрофилов, Т-лимфоцитов и макрофагов. Он препятствует васкуляризации опухолей и угнетает рост раковых клеток.
Некоторые интерлейкины активируют цитотоксическое противоопухолевое действие различных клеток иммунной системы и сдерживают рост ряда неоплазм. Это относится к ИЛ-4, ИЛ-7, ИЛ-9, ИЛ-12.
Ряд цитокинов, паракринно действующих в опухоли и вокруг неё, служат хемокинами и привлекают в очаг неоплазии клетки иммунной системы (ИЛ- 8 и ИЛ-1, ИЛ-6 и др.)
Паранеопластические явления
Паранеопластические явления– результат цитокиновых взаимодействий неоплазмы и организма и метаболических особенностей неоплазм.
Неопластические клетки влияют на организм своими секреторными продуктами, среди которых наибольшее значение имеют сигнальные молекулы, а также вносят нарушения в обмен веществ, удовлетворяя свои метаболические потребности. Активация иммунной системы приводит к появлению цитокинов, хроническое и системное действие которых характеризуется большим повреждающим потенциалом. Многие неоплазмы сами выделяют ферменты, гормоны, цитокины, а В-лимфомы даже антитела. Все это обусловливает возникновение комплекса паранеопластических феноменов:
Ø Раковая кахексия
Ø Мигрирующий тромбофлебит Труссо
Ø Паранеопластические остеопороз и гиперкальциемия
Ø Паранеопластическая анемия;
Ø Паранеопластическая пузырчатка
Ø Acanthosis nigricans
Ø Подагроидный синдром и другие.
Большинство из паранеопластических явлений не носит опухолеспецифичного характера (раковая кахексия, тромбофилитический синдром), но некоторые относительно специфичны (артериальная гипертензия при феохромоцитоме)
Неопластические клетки с их атипичным метаболизмом – ловушки глюкозы, аспарагиновой кислоты и биогенного азота. Этим и объясняется вредоносное действие неоплазмы, как «эндогенного колониального паразита», на организм носителя опухоли.
Преднеопластические заболевания
В связи с этой концепцией и многошаговым характером опухолевой трансформации, в практической онкологии сложились представления о преднеопластических (предраковых) заболеваниях, влияющие на принципы диспансеризации и профилактики. В группу преднеопластических относят все болезни и синдромы, при которых повышен риск последующей неопластической трансформации клонов клеток.
Это заболевания -с выраженным соматическим мутированием,
-с ослаблением иммунитета (особенно, клеточного)
-со снижением эффективности работы репаразных антимутационных механизмов
Возможно, при некоторых из таких болезней образуются эндогенные коканцерогены или канцерогены, а также аутоантитела, инактивирующие антионкобелки.
Примеры: Синдром Дауна повышает риск острого миелобластного лейкоза,
цирроз печени - рака печени,
болезнь Пэджета – предраковое заболевание для остеосаркомы,
дисплазия эпителия шейки матки - для рака шейки матки.
Понятия доброкачественной и злокачественнной опухоли
Принципиально важно, что опухоли, которые в клинике считаются доброкачествен-
ными, тоже являются неопластическими, а не гиперпластическими процессами.
Как правило, незрелые опухоли с инфильтрирующим ростом и дающие метастазы называются злокачественными. Зрелые опухоли с экспансивным ростом — доброкачественными.
Впрочем, некоторые доброкачественные опухоли обладают инвазивным (инфильтрирующим) ростом, хотя никогда не метастазируют (гемангиомы, лимфангиомы).
Злокачественные опухоли, как правило, не одоброкачествляются. Исключение:невробластома, трансформируемая в ганглионеврому. Приблизительно в 4-5% случаев выявляется остановка роста опухоли, она может «дозреть» до ганглионевромы, иногда самостоятельно, иногда при лечении.
Степень дифференцировки, в целом, коррелирует с доброкачественностью, но известно много исключений: так, феохромоцитома может быть доброкачественна при относительно большой степени клеточного атипизма.
Доброкачественность – относительная клинико-прогностическая, а не абсолютно-биологическая характеристика неоплазм. Неопластический клон может созревать, дифференцироваться и оставаться, тем не менее, злокачественным, так как не способен к апоптозу и потенциально губит пациента, вытесняя нормальные клоны и не замещая полностью их функций (пример – хронический лимфолейкоз).
82. Нарушение всасывания углеводов пищи, нарушение синтеза, депонирования и расщепления гликогена, транспорта углеводов в клетке и их усвоения. Дисахаридазная недостаточность. Галактоземия. Фруктозурия. Пентозурия. Гликогенозы. Агликогенозы.
- Нарушение всасывания углеводов. Дисахаридазная недостаточность. Галактоземия, фруктозурия, пентозурия.
Снндром мальдигестии - нарушение полостного пищеварения, расщепления крахмала до ди- и олигосахаридов (при панкреатической недостаточности). Ведущий симптом – амилорея.
Синдром мальабсорбции – нарушение пристеночного переваривания и всасывания углеводов в тонком кишечнике, относится к наследственным аутосомно-рецессивным ферментопатиям.
Причины: недостаток дисахаридаз (лактазы, мальтазы, инвертазы, трегалазы), натрий-зависимого ко-транспортера моносахаридов (действие ядов флоридзина, уабаина – создание экспериментальной модели глюкозо-галактозной мальабсорбции и эссенциальной почечной глюкозурии). Типовые симптомы: бродильный понос, эксикоз, вторичная гипотрофия.
Синдромы мальабсорбции могут также предполагать ферментный блок в межуточном обмене углеводов (накопление и токсическое действие некоторых промежуточных продуктов обмена углеводов):
| Название | Недостаток фермента | Накапливающийся продукт | Клиника |
| Галактоземия | Галактокиназа | Галактоза, галактитол | Катаракта |
| Галактозо-1-фосфат-уридил-трансфераза | Галактозо-1-фосфат | Классическая тяжелая галактоземия (катаракта, ЗПМР, полиорганное поражение) | |
| УДФ-галактозо-4-эпимераза | УДФ-галактоза, глюкозо-1-фосфат | Мягкое течение | |
| Непереносимость фруктозы | Фруктоза-1-фосфат-альдолаза | Фруктозо-1-фосфат | Торможение распада гликогена и последующее развитие гипогликемии, усиление мобилизации липидов, стеатоз печени, гиперурикемия. |
| Доброкачественная эссенциальная фруктозурия | Фруктокиназа | Фруктоза | Повышенное содержание фруктозы в крови и моче, ложный диагноз – сахарный диабет, гипогликемии нет. |
| Эссенциальная пентозурия | L-ксилулозо-редуктаза | Ксилулоза | Повышенное содержание ксилулозы в крови и моче, ложный диагноз – сахарный диабет, гипогликемии нет. |
- Транспорт углеводов в клетке и их усвоение
Переносчики глюкозы в клетку по градиенту концентрации – GLUT (1-5). Их работа контролируется гормонами, в первую очередь, инсулином (наиболее выражен ответ на инсулин у GLUT 4 мышечной и жировой ткани). Нарушения в работе транспортеров глюкозы, в том числе наследственные дефекты, лежат в основе некоторых генетических моделей ИНЗСД. В данном случае возможны пострецепторные дефекты на этапе сигнализации о переброске переносчиков к мембране, на стадиях циркуляции переносчиков в цитоплазме. У некоторых линий спонтанно диабетических мышей обнаружено снижение GLUT 2 в островковых В-клетках – ослабление секреции инсулина в ответ на повышение содержания глюкозы в крови.
После поступления в клетку с помощью переносчика глюкоза фосфорилируется под действием гексокиназы (во всех тканях) и глюкокиназы (в печени). Описан наследственный аутосомно-доминантный дефект глюкокиназы – воспроизводится картина ИНЗСД (диабет взрослых в юности или MODY).
Глюкокиназа и гексокиназа активируются инсулином, который конкурирует за глюкокиназу с глюкокортикоидами. Следовательно, реакция фосфорилирования хронически недостаточная при сахарном диабете и гиперкортицизме и временно ослаблена при стрессе.
- Гликогенозы и агликогенозы
Гликогенозы – группа наследственных нарушений накопления гликогена и/или его утилизации, создающих недостаточность на путях поддержания уровня глюкозы в крови (пчень) и/или генерации энергии (мышцы). По патогенезу они делятся на печеночные (Ia, Ib, III, IV, VIa), мышечные (V,VII) и смешанные (II).
| Тип | Дефект фермента | Избыток вещества | Клиника |
| Ia (фон Гирке) | Глюкозо-6-фосфатаза. Аутосомно-рецессивное. | Глюкозо-6-фосфата - активатор гликогенсинтетазы | Встречается чаще всего. Гепатомегалия. Почки увеличены Гипогликемия, усиление липолиза, гиперлипопротеинемия I или V типа, ацетонемия, метаболический ацидоз, ацетонурия. |
| Ib (генокопия фон Гирке) | Транслоказа глюкозо-6-фосфата в ЭПР | -//- | Редкое. -//- + нейтропения |
| II (Помпэ) | α-1,4-глюкозидаза. Аутосомно-рецессивное. | Секвестрация гликогена в лизосомах. | Нередкое. Наиболее злокачественная. Смерть в грудном возрасте. Кардиомегалия, СН. Слабость скелетных мышц. Увеличение языка. |
| III (Форбса-Кори) | Амило-1,6-глюкозидаза (деветвящий). Аутосомно-рецессивное. | Разветвленный гликоген | 25% случаев. Более доброкачественное. Гепатомегалия, М.б. гиперлипопротеинемия. Фиброз печени. |
| IV (Андерсена) | Гликозил-4,6-трансфераза (ветвящий). Аутосомно-рецессивное. | Линейный гликоген. | Крайне редкое. Лет.исход в 1 год жизни. Ранний цирроз. |
| V (Мак Арделя) | Фосфорилаза мыш. ткани. Аутосомно-рецессивное. | Нормальный гликоген | Наиболее редкоеы. При физ. нагрузке судороги, миоглобинурия, повышение сывороточной креатинфосфокиназы. |
| VI (Херса) | Фосфорилаза печени. Аутосомно-рецессивное. | Нормальный гликоген. | Редчайшее. По течению как I. Гипогликемия, гиперлипопротеинемия, ацидоз. |
| VIb | Киназа фосфорилазы b. Сцеплено с Х-хр. | -//- | -//- |
| VII (Таруи) | Фосфофруктокиназа мыш.ткани. Аутосомно-рецессивное. | Фруктозо-6-фосфат. | Как б.Мак-Ардля с умеренным течением. Умеренная ГА, провоцируемая окислителями. |
Агликогеноз / гликогеноз 0 – наследственный аутосомно-рецессивный дефицит УДФ-глюкозо-гликогентрансферазы (гликогенсинтетазы). В тканях отсутствуют отложения гликогена. Болезнь совместима с жизнью, пораженные дети нуждаются в частом кормлении, тенденция к утренней гипогликемии, кетоз с рвотой и судорогами, ЗПМР.
83. Гипогликемические состояния. Их виды и механизмы. Патогенез гипогликемической комы и ее проявления
Гипогликемия – синдром, развивающийся при понижении уровня глюкозы в крови ниже 3,8 ммоль/л.
Виды и причины:
I. Физиологическая
- тяжелая и длительная физическая нагрузка
- длительное умственное напряжение
- у женщин в период лактации
II. Патологическая
- Инсулиновая
- передозировка инсулина при лечении сахарного диабета
- инсулома
аденома островковых клеток поджелудочной железы.
Инсулома- причина органического гиперинсулинизма (болезнь Харриса).
чаще всего располагается в хвосте поджелудочной железы, под капсулой, но может быть кишечной, печеночной, бронхолегочной и др.
Независимо от уровня глюкозы продуцирует С-пептид, проинсулин, инсулин.
Патогенез:
резкое снижение глюкозы в крови – недостаточное снабжение головного мозга – прекоматозные расстройства в ЦНС: кора>подкорковые структуры>средний мозг>продолговатый мозг – гипогликемическая кома.
При физической нагрузке, предменструальных изменениях уровня эстрогенов, ночью (голод) секретируются катехоламины, глюкагон, кортизол – тревога, внутренняя дрожь, холодный пот, тахикардия, слабость, затем судороги.
Триада Уипла:
1. Уровень глюкозы ниже 2, 75 ммоль/л
2. Поведенческие нарушения
утренняя оглушенность, дезориентированность, потеря сознания натощак, сумеречные состояния сознания, дурашливость, пританцовывания, агрессивность
3. Купирование явлений сахаром/ инфузией глюкозы
Со временем прогрессируют гипогликемические энцефалопатия, полиневрит, вторичное полифагическое ожирение, ускоряется развитие атеросклероза.
- Недостаточность контринсулярных гормонов
Снижение секреции гормонов гипоталамо-гипофизарной – надпочечниковой системы. Склонны люди, испытывающие хронический стресс.
Гипогликемия развивается между приемами пищи.
Проявления: агрессивность при голоде. - Недостаточность расщепления гликогена
- гликогенозы
- агликогенозы
- печеночно-клеточная недостаточность (гепатиты хронические и острые)
- лейциноз (синдром Мак-Куори) - Алиментарная
- общее голодание
- углеводное голодание
- кишечная или энзимопатическая мальбсорбция углеводов - Глюкозурия
- отравление монойодацетатом, флоридзином
- почечная недостаточность
Нарушается реабсорбция, кишечное всасывание глюкозы. - Транзиторная у новорожденнных
Недостаточность механизмов регуляции углеводного обмена. - Инсулинзависимы сахарный диабет
- физическая нагрузка, голод, передозировка инсулином при лечении инсулином
- микроангиопатия
микроангиопатия сосудов портальной системы гипофиза – вторичный гипопитуитаризм – нарушение продукции контринсулярных гормонов – ослабление ответа на гипогликемию.
- нефросклероз ( феномен Зуброды-Дана)
нефросклероз – хроническая почечная недостаточность – удлинение времени циркуляции инулина в крови, снижение реабсорбции глюкозы.
Гипогликемическая кома
- Уровень глюкозы до 3 ммоль/л
Нервозность, тремор, потливость, тревога, чувство голода.
При прогрессировании: притупление чувствительности, дезориентация, галлюцинации (состояние схожее с алкогольным опьянением). - Уровень глюкозы 2,5-3 ммоль/л
снижение выработки АТФ в нейронах – нарушение работы K/Na Ca/Mg насосов – утрата ионных градиентов- деполяризация клеток ЦНС – клонические судороги
· Уровень глюкозы ниже 2,5 ммоль/л
Потеря сознания.
В тяжелых случаях могут наступать отек и некроз отдельных участков мозга.
84. Гипергликемические состояния. Классификация гипергликемии. Виды, этиология. Патогенные последствия острой и хронической гипергликемии. Гипергликемическая кома. Роль гликозилирования белков в патологии.
Гипергликемия – повышение содержания глюкозы в крови выше 120 мг/дл (6,1 мМ/л). Этиология гипергликемии может быть различной. Выделяют следующие виды гипергликемий:
- Алиментарная – временное повышение уровня глюкозы в крови при быстром поступлении в организм избытка легкоусвояемых углеводов.
- Стрессорная – отражает действие катехоламинов, глюкагона, глюкокортикоидов и вазопрессина и наступает при действии различных стрессоров, в том числе – психоэмоционального характера.
- Судорожная – наступает при конвульсиях мышц вследствие усиления в них гликогенолиза. Примером гипергликемии при судорожных состояниях служит повышение уровня глюкозы в крови при эпилептических припадках и столбняке.
- Эндокринные – развиваются при гиперпродукции контринсулярных гормонов. Гипергликемией сопровождаются синдром и болезнь Иценко-Кушинга и другие формы гиперкортицизма, гипертиреоз, гиперпаратиреоз, глюкагонома, феохромоцитома, акромегалия и гипофизарный гигантизм. Контринсулярные регуляторы могут вызывать гипергликемию, действуя на различные звенья углеводного обмена.
Гипергликемия при повышении содержания глюкозы выше 8 мМ/л, приводит к глюкозурии. При этом теряются ценные энергетические эквиваленты. Повышая осмотическую активность плазмы и мочи, гипергликемия ведёт к полиурии, жажде и полидипсии. Хроническая гипергликемия стимулирует неэнзиматическое гликирование внеклеточных белков и усиливает синтез полиоловых соединений в клетках. Это вызывает тяжёлые осложнения. Не любая гипергликемия, а лишь очень высокое содержание глюкозы в крови, вызывает опасное острое нарушение водно-солевого метаболизма и гипергликемическую кому.
Гипергликемическая кома.
Гипергликемическая (гиперосмолярная) кома встречается реже, чем диабетическая (кетоацидотическая). В большинстве случаев она возникает у больных с ИНСД старше пятидесяти лет и относительно редко бывает в детстком и юношеском возрасте. В 50% случаев она развивается у лиц с нераспознанным до того или плохо леченым диабетом, часто в связи со стрессом, травмой, болезнью, резкой дегидратацией организма.
Данное осложнение может быть спровоцировано инфузией белковых и углеводных растворов, лечением иммунодепрессантами и дилантином (фенитоином), диализом. Наиболее частая клиническая ситуация, приводящая к гиперосмолярой коме, - декомпенсированный ИНСД у пожилых больных, которые не могут себя обслужить и лишены надлежащего ухода.
Некетогенная гиперосмолярная гипергликемическая кома – не менее опасное осложнение сахарного диабета, чем кетоацидоз, дающее без надлежащего неотложного лечения почти 50% уровень смертности. Ключевое звено её патогенеза – гиперосмолярность. Показатель осмолярности внеклеточной жидкости увеличивается до 500 мосмоль/л. Это результат глюкозурического осмотического диуреза без достаточной компенсации потерь воды.
Патогенез гиперосмолярной комы зависит от следующих факторов:
1. Гипергликемия. К гиперосмолярной коме приводит лишь быстро нарастающий и достигающий очень высокого уровня избыток глюкозы в крови: 55-200 мМ/л (1000 – 3600 мг/дл).
2. Гипернатриемия, гиперхлоремия. Уменьшение выделения натрия и хлора с мочой обусловлено повышением секреции альдостерона (в ответ на дегидратационную гиповолемию), а также снижением почечного кровотока.
3. Высокое содержание остаточного азота, а том числе – мочевины (из-за ограничения диуреза), повышение содержания общего белка сыворотки – тоже вносят вклад в гиперосмолярность.
Гиперосмолярность à резко выраженная внутриклеточная дегидратация (концентрация глюкозы в межклеточной жидкости и плазме крови намного больше, чем в интрацеллюлярном отсеке; нарушение водно-электролитного равновесия в клетках мозга влечёт выраженную неврологическую симптоматику и тканевую гипоксию ЦНС с потерей сознания) à сгущение крови à возникновение множественных тромбозов и тромбоэмболия сосудов.
У многих больных формируется тромбо-геморрагический синдром, следует закупорка почечных клубочков, прогрессирует острая почечная недостаточность. Развивается всё более выраженная олигурия и даже полная анурия.
Гиперосмолярная кома развивается, как правило, в течение нескольких дней, вначале отмечается полиурия, затем быстро возникают другие симптомы:
- поверхностное частое дыхание, типа тахипноэ, без запаха ацетона;
- двусторонний спонтанный нистагм и мышечный гипертонус, снижение сухожильных рефлексов;
- затемнение сознания, вплоть до сопора и комы.
Ключевую роль в лечении играет инфузия гипотонических растворов. Малые дозы инсулина применяются для снижения гипергликемии. Это дополняется антиацидотической терапией и коррекцией калиевого баланса. 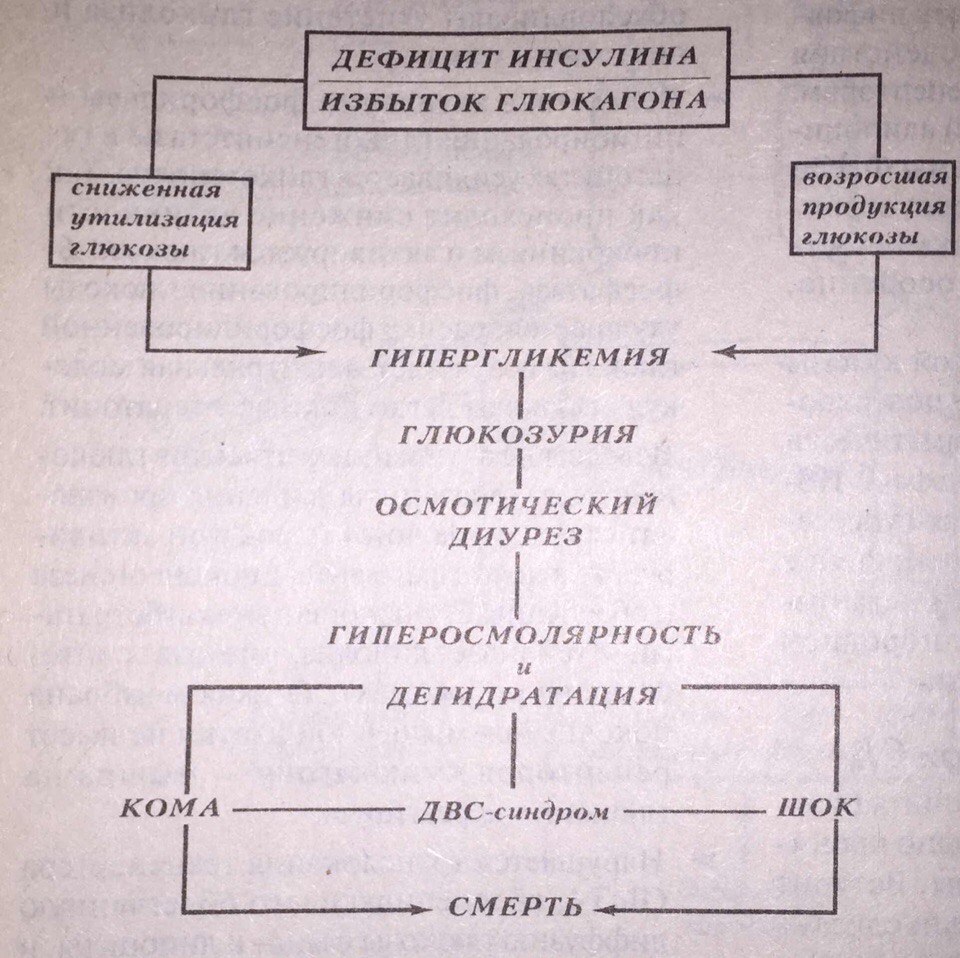
85. Сахарный диабет, его определение и классификация. Этиология и патогенез СД 1 типа. Работы Л. В. Соболева, Й. Меринга и О. Минковского. Экспериментальные модели СД, их виды и значение. Роль вирусов и аутоимунных процессов в поражении бета-клеток при СД 1 типа. Стадии течения СД 1 типа, их критерии.
Сахарный диабет – комплексное метаболическое заболевание, при котором все виды обмена веществ нарушаются из-за первичного поражения системы инсулиновой регуляции метаболизма.
Классификация
1. Первичный СД – идиопатическое расстройство механизмов инсулиновой регуляции метаболизма. Инсулина достаточно, чтобы не было кетоацидоза, но не поддерживает уровень глюкозы.
a. Первичный СД 1 типа (инсулинзависимый, гипоинсулинемический, юношеский)
1а – комбинация генетического и средового воздействия
1b – генетически обусловленные, со спонтанным аутоиммунным процессом, без явных признаков экзогенной провокации
1с – с первичным поражением β-клеток экзогенными диабетогенами (химическими, вирусами)
b. Первичный СД II типа (инсулиннезависимый, гиперинсулинемический, взрослых, тучных)
IIa – у нетучных больных
IIb – у тучных больных
IIc – в юношеском возрасте
2. Вторичный СД (диабетический/ гипергликемический синдром) – следствие других болезней, поражающих поджелудочную железу/систему регуляции углеводного обмена.
а. Вызванный неаутоиммунной деструкцией панкреатический β-клеток (хр. панкреатит и др.)
b. Вызванный эндокринными растройствами с гиперпродукцией контринсулярных гормонов (синдром Кушинга и др.)
с. Ятрогенный диабет при применение медикаментов (АКТГ и др.)
d. При генетически обусловленных синдромах (липидодистрофии)
Этиология СД 1: генетическая предрасположенность + ряд экзогенных факторов (вирусы –Коксаки, краснухи, реовирус, энтеровирус, эпидемический паротит, цитомегаловирус, Эпштейна-Барр и химические диабетогены – бычий сывороточный альбумин; Вакор – для борьбы с грузынами; Аллоксан, стрептозоцин; пентамицин – для лечения пневмоцистоза; Нитрозамины – в копченных продуктах)  аутоимунный процесс цитолиз островков β-клеток.
аутоимунный процесс цитолиз островков β-клеток.
Патогенез СД 1: прогрессирующая гибель β-клеток изменение гетерочиклических отношений в островках (инсулин, глюкагон, соматостатин)  инсулинопения и избыток контринсулярных гормонов нарушение утилизации глюкозы нарушение всех видов метаболизма хронизация нарушений осложнения (главное – микроангиопатия) + подавление пролиферации β-клеток антиклеточными АТ и медиаторами аутоиммунного воспаления.
инсулинопения и избыток контринсулярных гормонов нарушение утилизации глюкозы нарушение всех видов метаболизма хронизация нарушений осложнения (главное – микроангиопатия) + подавление пролиферации β-клеток антиклеточными АТ и медиаторами аутоиммунного воспаления.
Экспериментальные модели СД 1:
1. Хирургические - Панкреатэктомические:
На собаках: (Меринг, Минковский) – после операции у животных типичная картина ИЗСД с истощением, кетоацидозом и гибелью. Минковский - пересадка кусочка поджелудочной железы под кожу собаки, перед операцией, предохраняет ее от СД. Соболев – аутолиз экзокринной части поджелудочной железы, перевязывая выводной проток нет СД вещество, нехватка которого вызывает СД, вырабатывается островками Ларгенганса + рекомендации по его получению перевязкой выводных протоков у новорожденных телят (экзокринная часть слабо развита) Батинг и Бест выделение инсулина.
На южноамериканских жабах: Усай – удаление панкреатической железы + удаление аденогипофиза + надпочечника СД значительно ослабевает; предварительная гипофизэктомия резистентность к панкреатоэктомии локализация продукции контринсулярных гормонов. Субтотальная резекция щитовидки (удаляют часть) латентный сахарный диабет (переходит в явный при перекармливании углеводами/нагрузка контринсулярными гормонами (тироидные, соматотропин, глюкокортикоиды).
Дата добавления: 2019-03-09; просмотров: 819; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!