Гемолитическая, микросфероцитарная анемия.
Гемолитическая микросфероцитарная анемия (врожденная гемолитическая желтуха) является заболеванием, наследуемым по аутосомно-доминантному признаку (у 20 % больных наблюдаются спорадические случаи болезни). Заболевание связано с дефектом строения мембраны эритроцитов.
Мембрана становится хорошо проницаемой для натрия, что приводит к увеличению осмотического давления внутри эритроцита, и он приобретает сферическую форму, становится более хрупким. Неполноценные эритроциты захватываются и подвергаются быстрому разрушению селезеночной тканью, развивается гемолитическая анемия. Существует также мнение, что при этом виде анемии селезенка продуцирует избыточное количество аутогемолизинов. За счет гиперфункции селезенки возникает спленомегалия.
Клиническая картина и диагностика. Заболевание начинается в раннем возрасте, а иногда с момента рождения. Оно может протекать с развитием гемолитических кризов, при которых наблюдается быстрое (в течение нескольких дней) нарастание анемии и клинических проявлений гемолитической желтухи. В этот период у больного возникают
-тошнота
-рвота
-боли в верхней части живота
-тахикардия
-одышка
-гипертермия
-бледность кожных покровов
-которая быстро сменяется нарастающей желтухой.
У детей подобные кризы могут привести к смерти. Нередко гемолитические кризы провоцируются острыми инфекционными заболеваниями.
|
|
|
При бескризовом течении на первый план выступают симптомы анемии и гемолитической желтухи. Анемия весьма умеренная, желтуха редко бывает интенсивной. В этих случаях заболевание может впервые отчетливо проявиться в подростковом, юношеском и даже зрелом возрасте.
В связи с повышением уровня билирубина в крови у 50—60 % больных с гемолитической микросфероцитарной анемией возникает желчнокаменная болезнь (приступы печеночной колики, острого или первично-хронического холецистита). Поэтому наличие конкрементов в желчном пузыре у детей, особенно младше 10 лет, является показанием к тщательному обследованию для выявления гемолитической микросфероцитарной анемии.
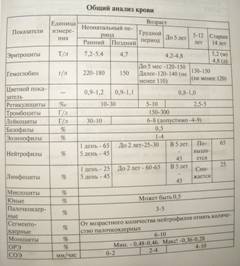
При физикальном обследовании определяют увеличенную селезенку. Печень, как правило, не увеличена. Ведущим методом диагностики является лабораторное исследование крови, при котором обнаруживают микросфероцитоз, снижение осмотической стойкости эритроцитов. В норме эритроциты начинают разрушаться в 0,47 % растворе натрия хлорида, при гемолитической анемии этот процесс начинается уже в 0,6 % растворе натрия хлорида. В общем анализе крови отмечают ретикулоцитоз. При исследовании мазка костного мозга выявляют гиперплазию красного ростка. Во время криза наблюдают ретикулоцитопению и гипоплазию красного ростка костного мозга. После криза число ретикулоцитов существенно увеличивается. Характерными лабораторными признаками гемолитической анемии являются анемия, увеличение концентрации непрямого билирубина в крови, повышенное содержание уробилина в моче и стеркобилина в кале.
|
|
|
Лечение. Консервативное лечение дает временный эффект и не предупреждает дальнейшего прогрессирования болезни и развития гемолитического криза. Радикальным способом лечения гемолитической микросфероцитарной анемии является спленэктомия. Хотя основная причина болезни кроется не в гиперплазии селезенки и повышении ее функции, а в изменении эритроцитов, спленэктомия приносит больному полное выздоровление. В результате этой операции длительность жизни эритроцитов (даже неполноценных, аномальных) существенно увеличивается, исчезают анемия и желтуха. Если во время операции выявлены конкременты в желчном пузыре, то следует дополнить спленэктомию холецистэктомией (если позволяет общее состояние пациента). Оперативное вмешательство целесообразно выполнять в период ремиссии болезни, а у детей — в возрасте 3—4 лет.
|
|
|
Гемоглобинопатии.
Гемоглобинопатия — наследственное или врождённое изменение или нарушение структуры белка гемоглобина, обычно приводящее к клинически или лабораторно наблюдаемым изменениям в его кислород-транспортирующей функции либо в строении и функции эритроцитов.
СЕРПОВИДНО-КЛЕТОЧНАЯ АНЕМИЯ (дрепаноцитоз) - это наследственная гемоглобинопатия, связанная с таким нарушением строения белка гемоглобина, при котором он приобретает особое кристаллическое строение — так называемый гемоглобин S. Характеризуется умеренно выраженной хронической гемолитической анемией, рецидивирующими острыми болевыми кризами и повышенной восприимчивостью к инфекционным заболеваниям (главным образом Streptococcus pneumoniae). Серповидно-клеточная анемия наследуется по аутосомно-рецессивному типу.
Этиология:
На молекулярном уровне: дефект гена НВВ. В венозном русле HbS полимеризуется с формированием длинных цепей, изменяющих форму эритроцитов (становятся серповидными)
На клеточном уровне: Серповидные эритроциты вызывают увеличение вязкости крови, стаз; создают механическую преграду в мелких артериолах и капиллярах, приводя к тканевой ишемии (с чем связаны болевые кризы). Кроме того, серповидные эритроциты менее устойчивы к механическим воздействиям, что приводит к их гемолизу.
|
|
|
Клиническая картина:
* Усталость и анемия
* Приступы боли
* Отек и воспаление пальцев рук и/или ног и артрит
* Бактериальные инфекции
* Тромбоз крови в селезенке и печени
* Легочные и сердечные травмы
* Язвы на ногах
* Асептический некроз
* Повреждение глаз
* отставание в физическом развитии (особенно у мальчиков)
* спле-номегалия,
* холелитиаз
Диагностика: обнаружение полимеризо-ванного HbS при электрофорезе Нb
Лечение: при острых болевых кризах средней степени тяжести в амбулаторных условиях - ненаркотические анальге-тики (ибупрофен, парацетамол). При острых болевых кризах в стационарных условиях - наркотические анальгетики парентерально. Следует как можно быстрее снизить дозу наркотического анальгетика, а затем перейти на приём ненаркотических анальгетиков, например парацетамола, ибупрофена
Необходимо учитывать возможность развития дегидратации и ацидоза. При развитии инфекции до получения результатов бактериологического исследования следует назначить антибиотик, активный в отношении Streptococcus pneumoniae и Haemophilus influenzae
Поддерживающая терапия - трансфузии отмытых или размороженных эритроцитов, а также антикоагулянтов Трансплантация костного мозга.
Осложнения: Асептический некроз головки бедренной кости и другие некротические осложнения. Сепсис.
Цереброваскулярные расстройства с неврологическими проявлениями, Кардиомегалия, Ретинопатия, Гемосидероз.
Течение и прогноз:Анемия радикально неизлечима. На втором десятилетии жизни количество кризов уменьшается, но осложнения возникают более часто. Некоторые пациенты умирают в детстве от некротических осложнений или сепсиса. Большинство больных доживают в среднем до 50 лет.
ТАЛАССЕМИЯ — группа наследственных гемоглобинопатий, в основе которых лежит снижение синтеза полипептидных цепей, входящих в структуру нормального гемоглобина А.
Этиология: Нарушается синтез одной из четырёх цепей глобина. Причины повышенной гибели эритроцитов связаны с нарушенной структурой клетки из-за неправильного соотношения цепей глобина в гемоглобине. Кроме укорочения жизни эритроцитов при данном заболевании происходит гибель клеток предшественников эритроцитов в костном мозге.
• a-Талассемия вызвана дефектом синтеза a-цепи глобина. Содержание HbA2 и HbF обычно не увеличено
•• Делеции четырёх генов. Неспособность продуцировать ни одной a-глобиновой цепи приводит к избытку g-глобиновых цепей, образующих тетрамеры Hb, называемого Hb Барта. Hb Барта имеет сильное сродство к кислороду, что препятствует тканевому дыханию — развиваются тяжёлая анемия, сердечная недостаточность, гепато- и спленомегалия, генерализованный отёк, и наступает внутриутробная смерть в результате водянки плода
•• Делеции трёх генов (болезнь HbН, гемоглобинопатия Н). Несмотря на наличие выраженной анемии и повышенного содержания Hb Барта, образуется достаточное для развития плода количество a-глобина. В течение всей жизни у пациента сохраняется анемия, варьирующая от средней до тяжёлой. В постнатальном периоде преобладает HbH
•• Делеции двух генов (малая талассемия). Умеренная гипохромная микроцитарная анемия.
•• Делеция одного гена (состояние здорового носительства). Характерна нормальная картина периферической крови, включая нормальные концентрации Hb, Ht и количество эритроцитов. Патологию выявляют при количественном измерении глобиновых цепей или при анализе генома. Носитель может перенести обострение болезни HbH или малой талассемии.
• b-Талассемия (более 90% всех талассемий) развивается в результате экспрессии аномальных генов b-глобиновой цепи. Так как в геноме два аллеля b-глобина, существует две различные формы b-талассемии
•• Гомозиготная b-талассемия (большая талассемия, анемия Кули) — тяжёлое заболевание, проявляющееся в детском возрасте и заканчивающееся летально к 20 годам; пациенты обычно трансфузиозависимы. Клинически проявляется задержкой роста, гепатоспленомегалией, незначительной желтухой, гиперплазией костного мозга и деформацией костей. Содержание HbA2 снижено или повышено, HbF — значительно повышено
•• Гетерозиготная b-талассемия (малая талассемия) — обычно умеренная анемия, больные не зависят от трансфузий. Малая талассемия распространена в Италии и Греции. Содержание HbA2 повышено, HbF в норме или слегка повышено.
Диагностика:
1) микроцитоз,
2) гипохромия
3) пойкилоцитоз (эллиптоцитоз)
4) Малая b-талассемия — увеличение концентрации HbА2 (5% по сравнению 2,5% в норме)
5) Большая b-талассемия — значительное увеличение фракции HbF.
6) увеличение селезёнки, деформации черепа
7) нормальное или повышенное содержание сывороточного железа.
Лечение:
1) коррекция анемии с помощью переливания эритроцитов (удержание гемоглобина на уровне 85 г/л, выводя при этом избыток железа десфералом)
2) Ранняя трансплантация костного мозга.
3) по показаниям — спленэктомию.
Гемофилия.
Наследственные заболевания, обусловленные дефицитом или моллекулярными аномалиями одного из прокоагулянтов, учавствующих в активации свертывания крови.
Классификация.
Гемофилия А – дефицит 8 фактора свертывания крови,
гемофилия В – дефицит 9 фактора,
гемофилия С – дефицит 11 фактора (плазменного предшественника тромбопластина-РТА, б-нь Розенталя).
Гемофилия А и В наследуются по рецессивному сцепленному с X-хромосомой типу, болеют только мужчины, женщины – кондукторы гемофилии.
Гемофилия С наследуется аутосомно, болеют и мужчины, и женщины.
По степени дефицита выделяют:
1) тяжелую форму – с уровнем прокоагулянта ниже 2%,
2) средней тяжести – 2-5%,
3) легкую – выше 5%.
Клиника. Кровоизлияния в крупные суставы конечностей. Наиболее часто поражаются коленные суставы. Возникновение гемартроза. Длительные, рецидивирующие кр-течения возникают после травм и операций.
Диагностика. Увеличение времени свертывания крови, времени рекальцификации плазмы, снижение потребления протромбина в процессе свертывания крови. Дифференциацию форм гемофилии проводят также с помощью замены плазмы больного в указанных пробах тест-плазмой больного с заведомо тяжелой формой гемофилии А,В,С. Критерий диагноза – содержание отдельных прокоагулянтов.
Лечение:
Гемофилия А – антигемофильный глобулин А (VIII фактор), очищенные концентраты его, свежезамороженная плазма и криопреципитат (белковый препарат изогенной плазмы). Повторные трансфузии проводят с интервалом 8-12 часов. При острой геморрагической анемии – эрмасса, для достижения гемостаза при жел-киш кр/течениях – криопреципитат. Наряду с заместительной терапией – гемостатические препараты неспецифического действия (гемофобин, аминокапроновая к-та – противопоказана при почечных кровотечениях).
Гемофилия В – фактор Кристмана (IX фактор), очищенные концентраты его, препараты рекомбинантного фактора IX, концентраты факторов протромбинового комплекса, замороженная плазма.
Гемофилия С – трансфузионная терапия только при травмах и хирургических вмешательствах.
Пурпура.
Тромбоцитопеническая пурпура – заболевание, характеризующееся геморрагическими проявлениями в виде кровоизлияний под кожу и кровотечений, возникающих в результате снижения количества тромбоцитов.
Классификация.
По характеру иммунных нарушений:
-изоиммунные
-трансиммунные
-гетероиммунные (острая форма)
-аутоиммунные (хр форма)
по механизму развития:
-обусловленные нарушением продукции тромбоцитов
-вызванные нарушением распределения тромбоцитов
-вследствие повышенного разрушения.
По Шабалову (1982):
-первичные тромбоцитопенические пурпуры (идиопатическая, изоиммунные и трансиммунные тромбоцитопении, а также наследственные формы)
-вторичные симптоматические тромбоцитопении.
По течению:
-острая (до 6 мес)
-хроническая (с редкими рецидивами и непрерывнорецидивирующая).
Идиопатическая тромбоцитопеническая пурпура (ИТП, б-нь Верльгофа) объединяет аутоиммунные формы заболевания, причину аутоагрессии при которых выявить не удается.
Этиология: Срыв иммунологической толерантности к собственному антигену. В анамнезе можно выявить вирусную, реже бактериальную инфекцию, перенесенную за 4-6 недель до манифестации геморрагического синдрома. Болеют чаще девочки. Чаще заболевание начинается исподволь и носит хронический рецидивирующий или затяжной характер.
Клиника. Кровотечения из слизистых оболочек и петехии, хар-ны профузные кр/течения из носа, десен, обильные и длительные менструации. Особенности кожных геморрагий – возникновение без видимых причин, мелкоточечный характер, образование небольших синяков. Цвет меняется от пурпурно-красного (свежие экхимозы) до голубого, зеленоватого и, наконец, желтого. Локализация на передней поверхности туловища, верхних и нижних конечностях и особенно в местах, подверженных трению и сдавлению. Более крупные кр/излияния образуются в местах инъекций. Период выраженной кр/точивости сменяется периодом относительного клинического благополучия, острые формы – внезапное начало, бурное развитие геморрагического синдрома, тяжелое течение и полное выздоровление в теч 6 мес.
Диагностика. Резкое снижение Тр (ниже 100*10/л), кровоточивость развивается при 50 и ниже. В мазке крови – тромбоциты больших размеров, пойкилоцитоз пластинок (выход в кровь молодых форм). Резкое уменьшение продолжительности жизни Тр, увеличение времени кровотечения (по Дьюку до 15мин ибольше, норма 3-5мин). Положительный с-м жгута, время свертывания в норме, гепариновое время плазмы увеличено. Тромбоэластограмма – замедление времени р-ции и времени образования сгустка, значительно уменьшена максимальная амплитуда. В периферической крови – м.б. острая постгеморрагич анемия с нейтрофильным лейкоцитозом, при частых повторных кр/течениях – хр постгеморрагическая железодефицитная анемия. В костном мозге – гиперплазия мегакариоцитарного аппарата или норма. Иногда обнаруживают антитромбоцитарные антитела (иммуноферментные и –флюоресцентные методы).
Лечение. В период выраженных геморрагических проявлений – стероидные гормоны (преднизолон 1мг/кг до 2-3мг/кг + препараты К, альмагель или викалин при пероральном применении). При недостаточной эффективности гормонов назначают иммуномодуляторы (декарис 2,5мг/кг 2р/нед, 2мес, т-активин, тималин, делагил). При неэффективности консервативной терапии через 4-6 мес – спленэктомия. Иногда в период рецидива уменьшается геморрагический синдром, но Тр не увеличиваются, тогда стероидные гормоны + иммуносупрессанты (винкристин 1-2мг/м2 1р/нед 2мес, имуран, меркаптопурин, циклофосфан). Плазмоферез (удаление антител), с гемостатической целью – аминокапроновая к-та, ингибиторы протеаз, дицинон.
Лейкозы.
Острый лейкоз
Острый лейкоз — клональное (онкологическое) заболевание, первично возникающее в костном мозге в результате мутации стволовой клетки крови. Следствием мутации является потеря потомками мутировавшей клетки способности к дифференцировке до зрелых клеток крови. Морфологический субстрат оcтpыx лейкозов — бластные клетки.
Следствием мутации стволовой клетки является развитие в костном мозге клона клеток, утративших способность к созреванию. Неопластический клон вытесняет нормальные гемопоэтические клетки, что приводит к развитию дефицита зрелых клеток в периферической крови. Снижение количества или полное отсутствие зрелых клеток периферической крови обусловливает выпадение соответствующих функций периферической крови, что влечет за собой развитие клинических симптомов заболевания.
Лейкозные инфильтраты в виде диффузных или очаговых скоплений обнаруживаются в лимфатических узлах, селезенке и печени. Это приводит к увеличению размеров этих органов. В печени характерно развитие жировой дистрофии. Возможна лейкозная инфильтрация слизистых оболочек полости рта и ткани миндалин.
Развитие инфекционных осложнений происходит вслествие иммунодефицита, вызванного нарушением функции лейкоцитов. Чаще всего инфекционные осложнения имеют бактериальное происхождение, грибковые и вирусные инфекции встречаются реже. Могут развиться ангина, гингивит, стоматит, остеомиелиты челюстно-лицевой области, пневмония, бронхит, абсцессы, флегмоны, сепсис.
Геморрагический синдром при острых лейкозах обусловлен тромбоцитопенией, повреждением печени и стенок сосудов. Он проявляется геморрагическим диатезом петехиально-пятнистого типа. На коже и слизистых оболочках появляются «синячки» и петехии небольшого размера. Появление геморрагий легко провоцируется самыми незначительными воздействиями — трением одежды, легкими ушибами. Могут иметь место носовые кровотечения, кровотечения из десен, метроррагии, кровотечения из мочевыводящих путей. Геморрагический синдром может привести к весьма опасным осложнениям — кровоизлияниям в головной мозг и желудочно-кишечным кровотечениям.
Анемический синдром проявляется в виде бледности, одышки, сердцебиения, сонливости.
Клиника: общая слабость, утомляемоять, неопределенная боль в костях, непостоянный субфебрилитет, увеличение периферических л/у, селезенки. Сопровождается – ОРЗ, ангина, стоматит, кровоточивость, выраженная интоксикация, высокая температура. Нарушение функции почек, желудка, кишок, половых и др органов, миокардиодистрофия, иммунодефицит, сепсис, пневмония.
Диагностика – острый период - снижение эритроцитов, тромбоцитов, лейкоциты - повышены, снижены или в норме. Лейкограмма: бластные клетки, лейкемический провал. Костный мозг - бластная метаплазия, редукция эритроидного, гранулоцитарного и мегакариоцитарного ростков.
Лечение. Антиметаболиты – антагонисты предшественников нуклеиновых к-т (меркаптопурин, метотрексат), антимитотические средства - блокируют митоз в стадии метафазы путем денатурации тубулина (винкристин, винбластин), алкилирующие соединения – нарушают синтез нуклеиновых к-т (циклофосфан, имфопуран), противоопухолевые антибиотики-антрациклины – подавляют синтез ДНК и РНК (рубомицин, фарморубицин, карминомицин, адриамицин), ферментные препараты – L-аспарагиназа, эпиподофиллотоксины – действуют на G2-фазу (вепезид, тенипозид).
Хронический лейкоз
Хронические лейкозы отличаются от острых дифференцировкой опухолевых клеток и более длительным стадийным течением.
Первая стадия (моноклоновая, доброкачественная) заболевания характеризуется присутствием одного клона опухолевых клеток, течет годами, относительно доброкачественно, хронически.
Вторая стадия (злокачественная) обусловлена появлением вторичных опухолевых клонов, характеризуется быстрым, злокачественным течением с появлением множества бластов и называется злокачественной, поликлоновой стадией, или стадией бластного криза. 80% больных хроническими лейкозами погибают в стадии бластного криза.
Этиология: воздействие высоких доз радиации при взрыве атомной бомбы повышает риск хронического миелоидного лейкоза, но не хронического лимфоцитарного лейкоза. Длительный контакт с гербицидами или пестицидами – риск хронического лимфоцитарного лейкоза. Высоковольтные линии передач фактор риска развития лейкоза.
Симптомы: повышенная утомляемость, слабость, потеря веса, повышение температуры и боли в костях.
Снижение числа нормальных лейкоцитов повышает риск инфекционных заболеваний. Уменьшение числа тромбоцитов сопровождается кровоизлияниями, кровотечениями из носа и десен. Распространение лейкоза из костного мозга в другие органы и ЦНС приводит к головной боли, слабости, судорогам, рвоте, нарушению зрения. Увеличение лимфатических узлов, печени и селезенки.
Лечение ХЛЛ: Химиоптерапия хлорамбуцил, циклофосфамид. Лейкаферез (удаление избытка лейкоцитов, включая опухолевые клетки). В редких случаях трансплантация ствол. кл.
Профилактика инфекций
Лейкемоидные реакции.
ЛЕЙКЕМОИДНЫЕ РЕАКЦИИ - изменения в крови и органах кроветворения, напоминающие лейкозы и другие опухоли кроветворной системы, но всегда имеющие реактивный характер и не трансформирующиеся в ту опухоль, на которую они похожи.
ОТЛИЧИЯ ОТ ЛЕЙКОЗОВ:
1. Лейкемоидные реакции не являются самостоятельным заболеванием, а носят вторичный симптоматический характер, причем нередко очевидна причина, индуцировавшая развитие лейкемоидной реакции
2. Как правило, лейкемоидные реакции возникают вследствие воздействия на организм бактериальных, вирусных инфекций, чрезвычайных стрессорных раздражителей, а также разнообразных патогенных факторов бактериальной и небактериальной природы, вызывающих сенсибилизацию организма
3. С устранением действия основного причинного фактора возникает и быстрая нормализация состава периферической крови.
4. Для лейкемоидной реакции нехарактерны признаки опухолевой прогрессии, свойственные лейкозам, в связи с чем при них не возникают анемии и тромбоцитопении метапластического характера
5. Количество бластных элементов в периферической крови не превышает 1-2%.
Патогенез: в одних случаях - выход в кровь незрелых клеточных элементов, в других-повышенная продукция клеток крови либо ограничение выхода клеток в ткани, либо наличие нескольких механизмов одновременно.
Различают лейкемоидные реакции миелоидного, эозинофильного, лимфатического, моноцитарного, моноцитарно-лимфатического типов, а также вторичные эритроцитозы и реактивные тромбоцитозы.
Лейкемоидные реакции миелоидного типа наблюдаются при различных инфекционных и неинфекционньгх процессах, септических состояниях, интоксикациях эндогенного и экзогенного происхождения, тяжелых травмах, остром гемолизе. Среди реакций миелоидного типа доминируют промиелоцитарные нейтрофильные. Резкий промиелоцитарный сдвиг в пунктате костного мозга на фоне омоложения гранулоцитов крови может быть обусловлен токсикоинфекцией, аллергическими реакциями лекарственного происхождения, при выходе из иммунного агранулоцитоза.
Миелоидные лейкемоидные реакции развиваются на фоне тяжелых инфекционных и других заболеваний с повышенной или высокой температурой, иногда может быть увеличение селезенки.
Эозинофильные реакции крови сопровождают аллергические диатезы, сенсибилизацию организма паразитами, медикаментами, изредка опухолевый рост (например, лимфогранулематоз, Т-клеточную лимфосаркому, рак и т. п.). Нередко лейкемоидные реакции эозинофильного типа наблюдаются при токсокарозе (глистная инвазия, вызванная паразитами токсокарами), лямблиозе, миграции личинок аскарид.
Лейкемоидные реакции лимфатического типа чаще всего являются результатом вирусной инфекции. Наиболее распространенный реактивный лимфоцитоз - малосимптомный инфекционный лимфоцитоз. По картине крови его легко можно принять за хронический лимфолейкоз, но он встречается почти исключительно у детей, а у них почти не бывает хронического лимфолейкоза. Инфекционный лимфоцитоз продолжается обычно несколько дней, сопровождается легкими катаральными явлениями. Реактивный лимфоцитоз может возникать после спленэктомии.
Лейкемоидные реакции моноцитарного типа встречаются при туберкулезе, саркоидозе, макроглобулинемии Вальденстрема, хронических воспалительных процессах. У детей такой тип реакции лейкоцитарного ростка костного мозга встречается крайне редко. Реактивный моноцитоз (повышение числа моноцитов) отличается от хронического моноцитарного лейкоза наличием признаков какого-либо заболевания, в то время как хронический моноцитарный лейкоз в течение первых лет болезни практически бессимптомен. В сомнительных случаях при длительно наблюдаемом моноцитозе показана трепанобиопсия костного мозга. При реактивном моноцитозе костный мозг нормален.
Диатезы.
К геморрагическим диатезам относят заболевания, в основе которых лежат нарушения сосудистой стенки и различных звеньев системы гемостаза, обусловливающие повышенную кровоточивость или склонность к ее возникновению.
Этиология и патогенез
Патогенез наследственных геморрагических состояний определяется нарушением нормальных гемостатических процессов: аномалиями мегакариоцитов и тромбоцитов, дефицитом или дефектом плазменных факторов свертывания крови, неполноценностью мелких кровеносных сосудов. Приобретенные геморрагические диатезы обусловлены ДВС-синдромом, иммунными поражениями сосудистой стенки и тромбоцитов, токсикоинфекционными поражениями кровеносных сосудов, заболеваниями печени, воздействиями лекарственных средств.
Классификация
1. Геморрагические диатезы, обусловленные дефектом тромбоцитарного звена
— недостаточность количества тромбоцитов
— функциональная неполноценность тромбоцитов
— сочетание количественной и качественной патологии тромбоцитов
2. Геморрагические диатезы, обусловленные дефектом прокоагулянтов (гемофилии) — недостаточное их количество, необходимое для формирования фибрина
— недостаточная функциональная активность отдельных прокоагулянтов
— наличие в крови ингибиторов отдельных прокоагулянтов
3. Геморрагические диатезы, обусловленные дефектом сосудистой стенки
— врожденные
— приобретенные
4. Геморрагические диатезы, обусловленные избыточным фибринолизом
— эндогенным (первичным и вторичным)
— экзогенным
5. Геморрагические диатезы, обусловленные сочетанием нарушений различных компонентов системы гемостаза (болезнь Виллебранда, ДВС-синдром и пр.)
Геморрагический васкулит.
Геморрагический васкулит - иммунокомплексное заболевание, в основе – множественный микротромбоваскулит.
Этиология. Предшествует развитию стрептококковая или вирусная инф-я, медикаментозная, пищевая аллергия, эндогенная сенсибилизация.
Патогенез. в микрососудах – асептическое воспаление с глубокой деструкцией стенки, тромбообразование вследствие повреждающего действия циркулирующих низкомолекулярных иммунных комплексов и активированных компонентов системы комплемента.
Классификация. Кроме классической выделяют вторичные формы, наблюдающиеся при коллагенозах (ревматоидный артрит), лимфопролифе-ративных процессах, заболеваниях печени.
По локализации процесса: кожная, кожно-суставная, абдоминальная, почечная, смешанная формы.
По течению: молниеносная, острая, затяжная, рецидивирующая, хроническая, персистирующая формы.
Клиника. Кожные высыпания – мономорфная, возвышающаяся над поверхностью кожи, с четкими краями, красно-багрового цвета папулезно-геморрагическая сыпь, не исчезающая при надавливании (при тяжелом течении элементы сливаются с некрозом в центре). После разрешения сыпи остается пигментация. Сыпь возникает на нижних конечностя, в области суставов, реже - на ягодицах, туловище, верхних конечностях, на лице; кожные элементы различной степени зрелости. Суставной синдром – летучая боль различной интенсивности в голеностопных, коленных и др суставах, м.б. с кожными высыпаниями или после их возникновения. Абдоминальный синдром – у детей и стариков, приступы сильной боли в животе, рвота, мелена, свежая кровь в кале, при раздражении брюшины – с-мы острого живота. Почечный синдром – через некоторое время после начала заболевания по типу острого нефрита без портальной гипертензии.
Диагностика. В острый период – нейтрофильный лейкоцитоз, повышение СОЭ, содержания белков плазмы. При поражении почек – в моче белок, Эр, цилиндры. В тяжелых случаях – нарушение коагуляционного гомеостаза хар-ое для ДВС-синдрома.
Лечение. Постельный режим, диета (исключаются продукты, вызывающие аллергизацию). При наличии инфекции – малоаллергизирующие антибиотики (цепорин). Гепаринотерапия 300-400Ед/кг/сут в/в или п/к под контролем тромбинового времени и аутокоагуляцион теста. Достижение эффекта – увеличение показателей в 2 раза, нет эффекта – гепарин до 1000 Ед/кг/сут. Для стимуляции фибринолиза – никотиновая к-та, улучшение микроциркуляции – трентал. Нестероидные противовоспалительные – вольтарен, индометацин. Преднизолон 0,5-0,7мг/кг. При неэффективности – плазмоферез, иммунодепрессанты.
Врожденный гипотиреоз.
Врожденный гипотиреоз
Патогенетически гипотиреоз классифицируется:
1. Первичный (тиреогенный).
2. Вторичный (гипофизарный).
3. Третичный (гипоталамический) (казуистика).
Этиология: полная или частичная недостаточность тиреоидных гормонов.
В 85 - 90% случаев врожденный гипотиреоз - первичный и связан с дисгенезией щитовидной железы. При этом чаще всего имеется аплазия, гипоплазия или дистопия щитовидной железы.
В 5 - 10 % случаев первичный врожденный гипотиреоз обусловлен дисгормоногенезом: дефектом рецепторов ТТГ, дефектом транспорта йодидов, пероксидазной системы или синтеза тиреоглобулина. Этот вариант врожденного гипотиреоза часто носит аутосомно-рецессивнй характер и сопровождается увеличением щитовидной железы.
Врожденный гипотиреоз является вторичным или третичным (патология гипофиза и/или гипоталамуса) не более, чем в 3-4% случаев. Чрезвычайно редкой формой врожденного гипотиреоза является синдром периферической резистентности к тиреоидным гормонам, при котором уровни ТТГ и тиреоидных гормонов в пределах нормы.
Отдельно рассматривается транзиторный гипотиреоз новорожденных. Последний может быть связан с приемом беременной тиреостатических препаратов (пропицил, метказолил), быть индуцирован материнскими антителами к щитовидной железе. Чаще всего это состояние развивается у недоношенных и незрелых новорожденных, особенно в областях эндемичных по дефициту йода.
Клиника: в раннем постнатальном периоде:
1)переношенная беременность (более 40 недель)
2)большая масса тела при рождении
3)отечное лицо, губы веки, полуоткрытый рот с широким "распластанным" языком;
4)локализованные отеки в виде плотных "подушечек" в надключичных ямках, тыльных поверхностях кистей, стоп;
5)признаки незрелости при доношенной по сроку беременности.
6)низкий грубый голос при плаче, крике;
7)позднее отхождение мекония;
8)позднее отхождение пупочного канатика, плохая эпителизация пупочной ранки;
9)затянувшаяся желтуха.
На 3-4 месяце жизни:
1)сниженный аппетит, плохая прибавка в массе тала;
2)метеоризм, запоры;
3)сухость, бледность, шелушение кожных покровов;
4)гипотермия (холодные кисти, стопы);
5)ломкие, сухие, тусклые волосы;
6)мышечная гипотония.
После 5 - 6 месяца жизни:
нарастает задержка психомоторного и физического развития ребенка.
Диагностика: исследования уровней ТТГ и Т4. Кровь забирается путем чрезкожной пункции (чаще из пятки) на 4 - 5 день после рождения. У недоношенных детей оптимальным сроком взятия крови на ТТГ являются 7 - 14 сутки жизни. Если ТТГ менее 20 мкЕд/мл = вариант нормы, ТТГ > этого показателя все образцы крови должны быть проверены повторно. Уровень ТТГ >50 мкЕд/мл позволяет заподозрить гипотиреоз, а уровень > 100 мкЕд/мл - на наличие заболевания. Если при повторном обследовании пациентов с уровнем ТТГ 20 - 50 мкЕд/мл, уровень ТТГ > 20 мкЕд/мл, а уровень общего Т4 оказывается < 120 нмоль/л – то лечение.
Лечение: Назначение L-тироксина. Детям 1 года проводится уточнение диагноза путем двухнедельной отмены L-тироксина и исследования уровней ТТГ и Т4. При получении нормальных показателей лечение не возобновляется. Всем детям с врожденным гипотиреозом для установления этиологии заболевания необходимо провести УЗИ и/или радиоизотопное сканирование щитовидной железы. Заместительная терапия L-тироксином ведется в исходной дозе 25 - 50 мкг/сут или 8 - 12 мкг/кг/сут. При пересчете на площадь поверхности тела доза препарата у новорожденных составляет 150 - 200 мкг/м2, у детей старше года - 100 - 150 мкг/м2
Диффузный токсический зоб.
ДИФФУЗНЫЙ ТОКСИЧЕСКИЙ ЗОБ
Органоспецифическое а-имунное забол-е, с повыш продукцией тиреоидных гормонов щит железой (ЩЖ).
ЭТИОЛОГИЯ
Наследуется сцеплено с системой НLA. Дефицит Т-супрессоров – образование тиреоид-стимулирующих АТ - они конкурируют с ТТГ за рецепторы на тиреоцитах – гиперплазия железы, повыш продукции тиреогормонов.
ПАТОГЕНЕЗ
Избыток тиреоидных гормонов – катехоламиноподобный эффект - повыш нервную возбудимость. повыш теплопродукция, утилизация гл-зы, повыш глюконеогенез, липолиз. повыш анаболические и катаболические процессы – дистрофия миокарда, печени, мышеч слабость, относительная надпочечниковая недостаточность.
КЛИНИКА Обусловлена:
1) местным а-имунным процессом в щитовидке,
2) признаками гипертиреоза,
3) ассоциированными а-имунными заболеваниями.
ЩЖ диффузно увелич до 2-3 степени, плотноэластичная, над ней – сосудистые шумы. С-мы нарастают за 6-12 месяцов, ребенок возбудимый, раздражительный. повыщ двигательная активность, повыш аппетит, жажда. Б-ной худеет, резкая мышечная слабость, кожа бархатистая, горячая, влажная, потливость, пигментация в области век, повыш ЧСС в покое, расшир границы сердца влево, на верхушке и в т. Боткина сист шум. повыш пульсовое АД. Офтальмопатия – поражение ретробульбарной клетчатки и глазодвиг м-ц специфич аутоАТ, глазные щели расширены.
ДИАГНОЗ Т3 больше 3 нмоль/л, Т4 больше 200 нмоль/л, ттг меньше 0,1 мкЕД/мл
ДИФДИАГНОЗ
1) С ВСД – при ВСД тахикардия, эмоц-ное возбуждение, похудание имеют непостоянный хар-р,
2) с б-нью Пламмера, раком – определяются пальпаторно иил на сканограммах узлы,
3) гнойный, подострый тиреоидиты – быстрое увелич ЩЖ, её болезненность, гипертермия, лаб признаки воспаления,
4) при феохромоцитоме - АД обычно повышено и диаст-кое и систолическое, много катехоламинов. Тиреоидных – норма.
ЛЕЧЕНИЕ
1) консервативное – длительное применение тиреостатиков (мерказолил), контролем служит нормализация ТТГ,
2) хирургич – субтотальная струмэктомия – только после медикаментозного устранения тиреотоксикоза,
3) р-активный I – при неэффективности консервативного и невозможности оперативного,
4)при тяжелом тиреотокс-зе - анаприлин, обзидан, при сочетании с офтальмопатией 2-3 степени – гормоны.
Аутоиммунный тиреоидит.
Аутоиммунный тиреоидит – хроническое заболевание щитовидной железы, характеризующееся аутоиммунным повреждением ее клеток.
Более распространен у девочек и у детей с заболеваниями щитовидной железы в семейном анамнезе.
Развивается чаще в пре- и пубертатном периоде.
Этиология:
Генетически обусловленный дефект иммунокомпетентных клеток - срыв естественной толерантности.
Этот процесс может быть запущен:
перенесенной вирусной инфекцией
травмой щитовидной железы
операцией на щитовидной железе
длительным бесконтрольным приемом йодсодержащих препаратов
действием радиационного излучения.
Патогенез:
выработка АТ к тиреоглобулину, тиреоидной пероксидазе , Т3, Т4, рецепторам тиреотропного гормона на мембранах тиреоцитов - деструкция клеток ЩЖ и снижение ее функции — гипотиреоз - повышенный выброс ТТГ гипофизом
рост сохранившегося эпителия ЩЖ с последующей инфильтрацией его лимфоцитами, макрофагами, плазматическими клетками.
ФАЗЫ АИТ:
1) эутиреоидная;
2) гипертиреоидная;
3) гипотиреоидная.
Гипер:
Клинические проявления:
-тахикардия,
-изменение АД с увеличением пульсового давления
-потливость
-тремор век и пальцев вытянутых рук
Гипо:
Клинические проявления:
-брадикардия
-снижение АД
-зябкость
-холодные конечности
-сонливость
-меньшая подвижность ребенка
-снижение успеваемости в школе
-снижение памяти
-отставание в росте и развитии.
Диагностика:
1) Пальпация: ЩЖ увеличена; наличие плотной консистенции ЩЖ, ее неоднородность, узловатость, иногда легкая болезненность.
2) Исследование ультразвуковой картины ЩЖ: увеличение органа; неоднородность эхоструктуры с наличием гипоэхогенных участков, иногда сочетающихся с гиперэхогенными; нечеткость контуров ЩЖ.
3) Тонкоигольная биопсия подозрительных участков ЩЖ с последующим морфологическим исследованием.
4) исследование в сыворотке крови концентрации гормонов ЩЖ — тироксина (Т4), трийодтиронина (Т3), а также ТТГ и тироксинсвязывающего глобулина (ТСГ).
в гипертиреоидную фазу: повышение уровней Т4 и Т3 и снижение ТТГ и ТСГ.
в гипотиреоидную фазу: повышение ТТГ иногда при нормальном уровне Т4 и Т3 или при одновременном снижении уровня тиреоидных гормонов.
5) исследование уровня АТ к тиреоидной переоксидазе и к тиреоглобулину
Лечение:
1. препараты для коррекции иммунитета (Т-активин)
2. нестероидные противовоспалительные препараты:
индометацин
метиндол
вольтарен
3. антибактериальные препараты при наличии болезненности ЩЖ или очагов хронической инфекции, что практически наблюдается у всех детей с ХАТ.
В фазу гипертиреоза:
1. тиростатики (2-3 недели)
тиамазол,
мерказолил
2. малые дозы L-тироксина (аналог Т4) при достижении эутиреоидного состояния
3. бета-адреноблокаторы
В фазу гипотиреоза:
1. синтетические гормоны щитовидной железы:
левотироксин (L-тироксин)
лиотиронин (аналог Т3)
их комбинации, в чистом виде или с добавлением калия йодида.






Дата добавления: 2018-04-15; просмотров: 336; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!