Другие 5 (гиперцистинурия, гистилинурия, лизинурия, мальабсорбция триптофана и метионина) – субстрат – специфичные.
Все описанные состояния – аутосомно-рецессивные многогенные наследственные болезни.
Цистинурия (самая встречаемая и клинически важная). Цистин – малорастворим. Он формирует камни в почках, мочевом пузыре, уретре. Имеет три типа: 1 – нет выраженной гипераминоацидурии, 2 – мочевые симптомы + снижено кишечное всасывание, увеличена почечная экскреция орнитина, лизина, аргинина, 3 – нарушение почечной, интактной кишечной абсорбцией аминокислот. Нагрузка пищевая цистеином не ведет к ухудшению мочевых симптомов, так как эта аминокислота плохо всасывается в кишечнике. + наследственная гиперцистеинурия (изолированно нарушен транспорт цистеина).
Болезнь Хартнупа – нарушено всасывание и увеличена экскреция крупномолекулярных нейтральных аминокислот, вторично (не хватает триптофана) раз-ся нарушения синтеза НАД.
Дибазикаминоацидурии – не реабсорбируется аргинин, орнитин, лизин (но не цистеин – не хар-но камнеобразование). Нарушается цикл мочевины, гипераммониемия, задержка роста. Умственное отставание.
Мальабсорбция триптофана – ведет к усиленному образованию в кишечнике производных индола. Поражение нервной системы (нарушается кроветворение). Кальциноз почек.
Метиониновая мальабсорбция – умственное отставание, задержка роста, гипопротеинемические отеки, нарушенное образование меланина.
Лизинурия – судороги, нарушение психомоторного развития.
|
|
|
Гистидинурия – нарушения синтеза гемоглобина и функций ЦНС.
Оксопролинурия – не расщепляется пирролидонкарбоновая кислота (побочный продукт при отщеплении аминокилот, переносимых в клетки, от глутатионового транспортера.
Синдром Лоува – наследственное, сцепленное с Х-хромосомой. Генерализованный дефект транспорта аминокислот. +ацидоз, задержка физического и психомоторного развития, двусторонняя глаукома и катаракта. Гипотонус мышц, черепная дизморфия.
Синдром Фанкони – аминоазот возрастает в мочи в 30-40 раз. Канальцевый ацидоз, полиурия с выведением всех аминокислот.
Синдром Людера-Шелдона – аминоцидурия без поражения без поражения кишечноц абсорбции аминокислот, сопровождается фосфатурией.
Синдром Роули-Розенберга – задержка физического развития, гипоплазия мышц, поражение легких, правожелудочковая сердечная недостаточность, дефект почечной реарбсорбции всех аминокислот.
Прибретенные нарушения транспорта: отправления тяжелыми металлами. Эндогенное отравление происходит при болезни Коновалова-Вильсона.
При наследственных нарушениях активации и рецепции витамина D, так же нарушается реабсорбция ряда аминокислот, из-за ингибирующего действия парат-гормона на этот процесс при вторичном гиперпаратиреозе.
|
|
|
До 3-х месяцев жизни система переноса еще не развивается в полную силу, есть место физиологической преходящей аминоацидурии новорожденных.
Аминоацидурия – 99% аминокислот должны реабсорбироваться тубулярным аппаратом почек. Следовательно, аминоацидурией или гипераминоацидурией называется такое состояние, когда с мочой выводится избыточное количество аминокислот и промежуточных продуктов их обмена.
Есть три группы основных механизмов возникновения данного состояния:
- повышается концентрация аминокислот в крови выше максимальных возможностей почечной реабсорбции (допустим, перекорм белками). В этой же группе – наследственные илиприобретенные нарушения процессов дезаминирования и переаминирования в печени.
- конкурентное ингибирование одной аминокислотой других аминокислот (такая аминоацидурия называется смешанной).
-дефект транспортера или сопряженного с ним энергетического процесса в самих канальцах.
Часто аминоацидурия сопровождается гипераминоацидемией.
Наследственные нарушения межуточного обмена аминокислот. 1Фенилкетонурии, 2алкаптонурия, 3лейциноз, 4гомоцистинурия, нарушение 5обмена тирозина. Этиология, патогенез, механизмы основных проявлений.
|
|
|
Наследственные нарушения межуточного обмена АК (аминоацидопатии) вызваны различными мутациями. В норме наиболее велика скорость обмена белков в нервной ткани. Поэтому наследственные аминоацидопатии - одна из причин слабоумия.
1 Фенилкетонурия (или фенилпировиноградная олигофрения) – мозаичная аминоацидопатия, мутация в 12 хромосоме, аутосомно-рециссивная, имеет несколько генокопий. В большинстве случаев сопровождается дефицитом печеночного фермента фенилаланин-4-гидроксилазы, что ведет к резкому увеличению фенилаланина в крови.
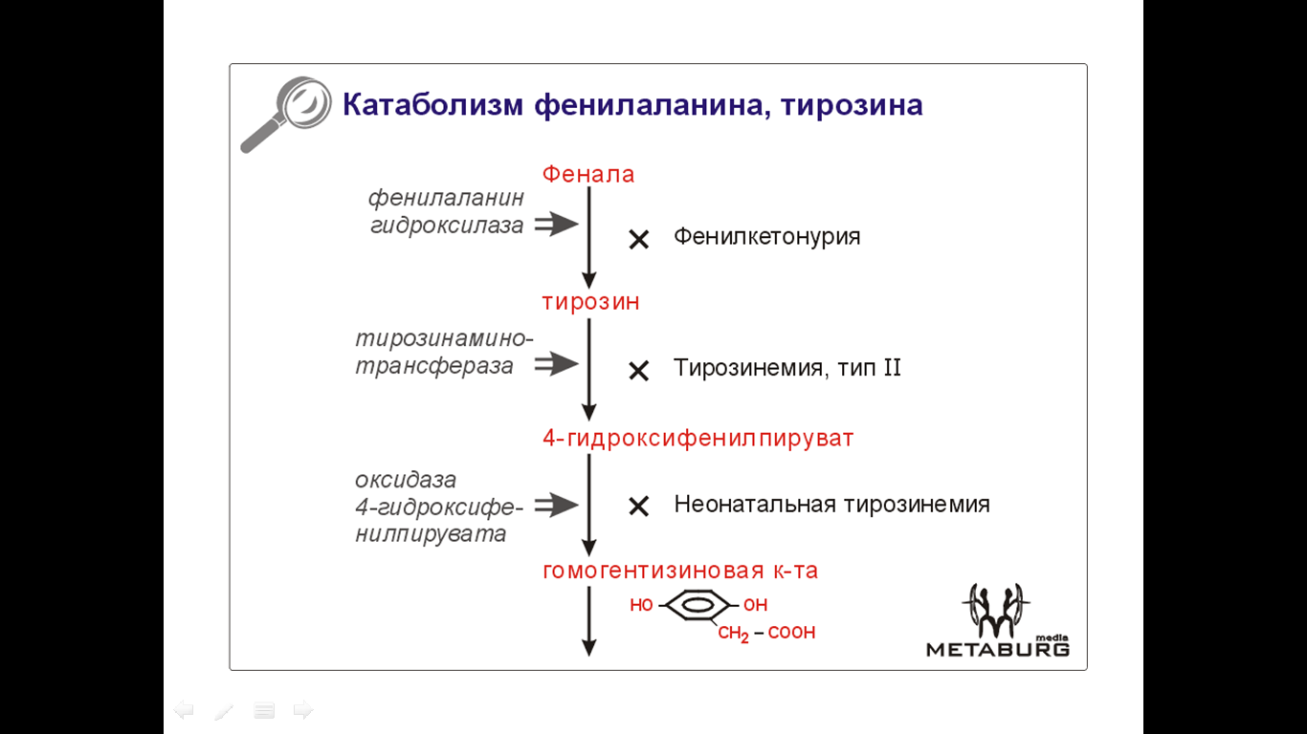
Недостаток превращения фенилаланина в тирозин, подавление избытком фенилаланина активности тирозиназы ведут к дефициту тирозиновых и триптофановых производных (меланина=>светлые кожа и глаза; катехоламинов=>гипотензия; серотонина=>судороги и тремор).
Избыток фенилаланина метаболизируется обходными путями, повышается концентрация продуктов его альтернативного метаболизма. Соответственно, в крови и моче повышается их концентрация. Это: фенилпировиноградная кислота, фенилмолочная кислота, фенилацетилглутамин. (Для диагностики используют реакцию Феллинга и тест Гатри.)
|
|
|
Другие продукты этого альтернативного метаболизма: фенилэтиламин, ортофенилуксусная кислота или фенилацетат. Эти соединения практически отсутствуют в норме и являются нейротоксинами, которые нарушают метаболизм липидов в мозге. В сочетании с дефицитом серотонина это ведет к прогрессирующему снижению интеллекта, выявляется через несколько месяцев после рождения. Задержка психомоторного развития столь глубока, что большинство больных детей не ходят (30%) и не говорят (2/3), и лишь в 4-5% слабоумие остается на уровне дебильности. У других – имбецильность или идиотизм.
Основной метод лечения – ограничение потребления фенилаланина с целью снизить его концентрацию в крови с 16мг/дл до 3-12мг/дл. Соблюдение диеты особенно важно до полового созревания и во время беременности (альтернативные метаболиты фенилаланина оказывают тератогенное действие).
Болезнь может быть вызвана и дефектом другого фермента – дигидроптеридинредуктазы. Она не лечится ограничением потребления фенилаланина, имеет худший прогноз.
2 Алкаптонурия – аутосомно-рецессивное заболевание. Причина – дефект оксидазы гомогентизиновой кислоты. Накопление гомогентизиновой кислоты ведет к ее превращению полифенолоксидазой в хиноновые полифенолы. Эти хиноновые полифенолы составляют «охронозный пигмент» (алкаптон), выводимый почками и обусловливающий потемнение мочи больных на воздухе и при подщелачивании. (ПАЦИЕНТЫ В БУКВАЛЬНОМ СМЫСЛЕ ВЫДЕЛЯЮТ ФОТОПРОЯВИТЕЛЬ: при попадании на фотобумагу подщелоченная моча больного вызывает ее почернение!)
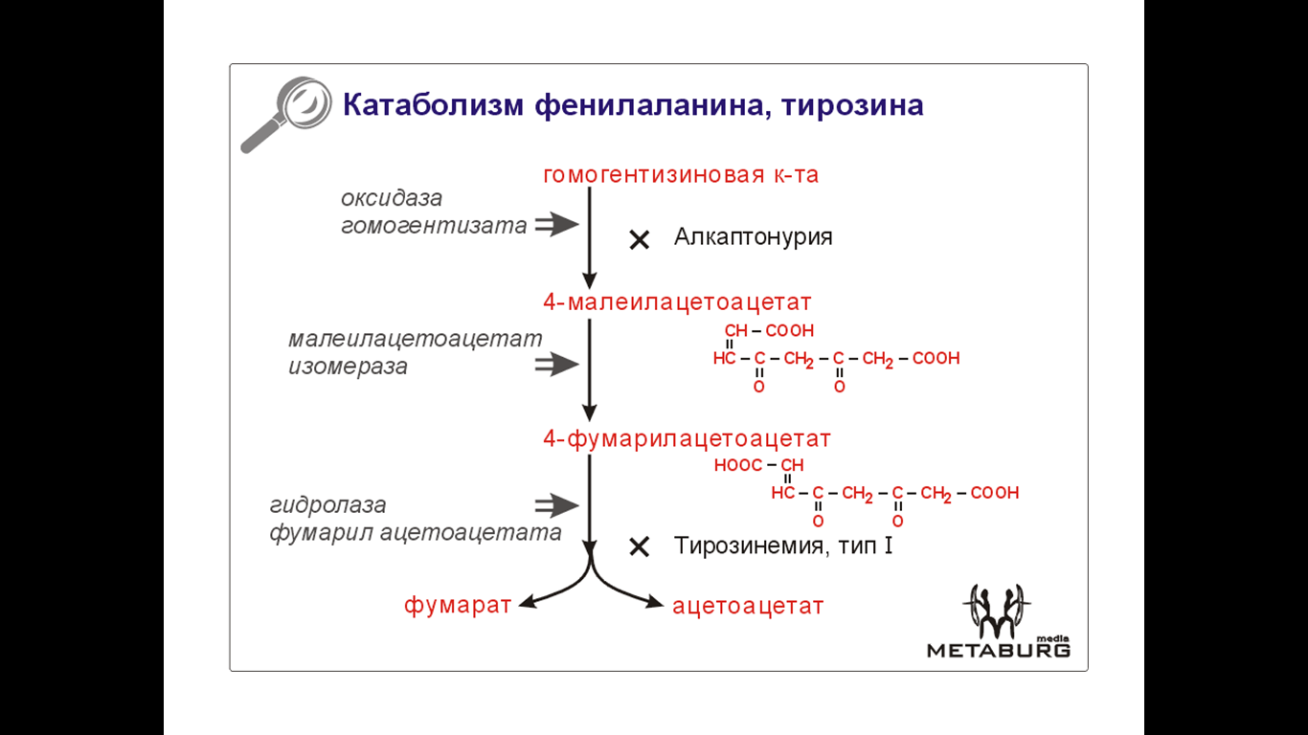
Охронозный пигмент экскретируется не полностью и откладывается в соединительно-тканных образованиях: хрящах (делает их хрупкими, что со временем вызывает кальцификацию и артрит), склере (может быть первым проявлением болезни).
Гомогентизиновая кислота (накапливаясь) ингибирует фермент лизилгидроксилазу, участвующую в синтезе коллагена.
Радикально болезнь не лечится. Можно лишь уменьшить степень хондропатии, защищая активность лизилгидроксилазы большими дозами аскорбиновой кислоты.
3 Лейциноз (разветвлено-цепочечная кетонурия) – аутосомно-рецессивное заболевание. При этой патологии нарушено окислительное декарбоксилирование (окисление) разветвленных кетокислот, которые синтезируются путем дезаминирования лейцина, изолейцина, валина.
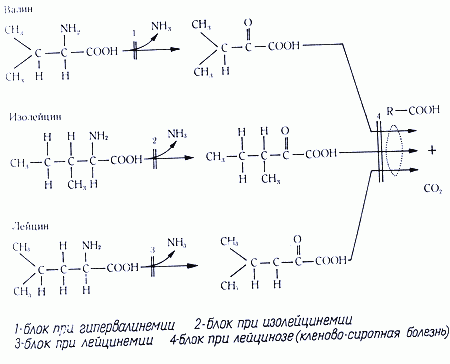
В результате накапливаются и кетокислоты, и вышеперечисленные аминокислоты. Наиболее патогенно накопление лейцина. Лейцин – единственно чисто кетогенная аминокислота, то есть в норме окисляемая до ацетоацетата и ацетилкоэнзима А. Из-за большой роли кетоновых тел в энергообеспечении мозга (особенно при гипогликемии), данная патология чревата развитием умственной отсталости, судорог, мышечной ригидности; также отмечаются гипогликемия, гипотония, больные могут впадать в летаргию, имеется кетоацидоз.
Нарушение обмена лейцина может быть связано с дефектом изовалерил-коА-дегидрогеназы - изовалератацидемия. При обеих болезнях нарушается превращение кетокислот в липиды, => нарушается продукция миелина, страдает энергетика мозга, уменьшается образование ГАМК.
Лечение сводится к специальной диете с ограничением потребления алифатических разветвленных АК. Без лечения – смерть за несколько месяцев.
4 Гомоцистинурия – синдром, имеющий у разных больных разную этиологию. В большинстве случаев причина в дефекте сериндегидратазы, при этом серин и гомоцистеин не могут с должной скоростью образовывать цистатионин. Накапливаются: гомоцистеин, гомоцистин, метионин.
Лечение – витамин В6, активирующий метаболизм гомоцистеина.
Другие дефекты, вызывающие гомоцистинурию – это нарушения в протекании Nметилтетрагидрофолат-гомоцистеин-метилтрансферазной реакции:

· Дефект синтеза апофермента
· Дефицит метилированной формы витамина В12
· Недостаточная активность N-метилентетрагидрофолат-редуктазы
В таких случаях: резистентность к терапии витамином В6; нет накопления метионина; развивается умственная отсталость, эктопия хрусталиков, остеопороз, сколиоз, патологические переломы.
При всех формах гомоцистинурии может быть тромбоэмболический синдром; возрастает риск атеросклероза.
5 Нарушения обмена тирозина:
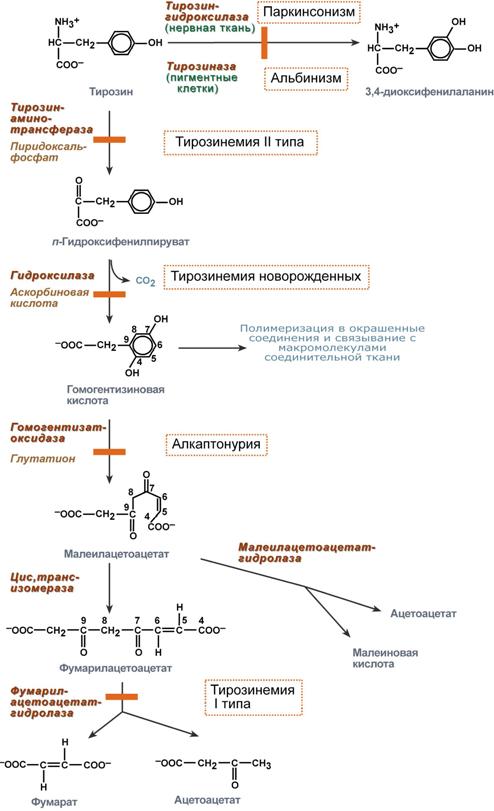
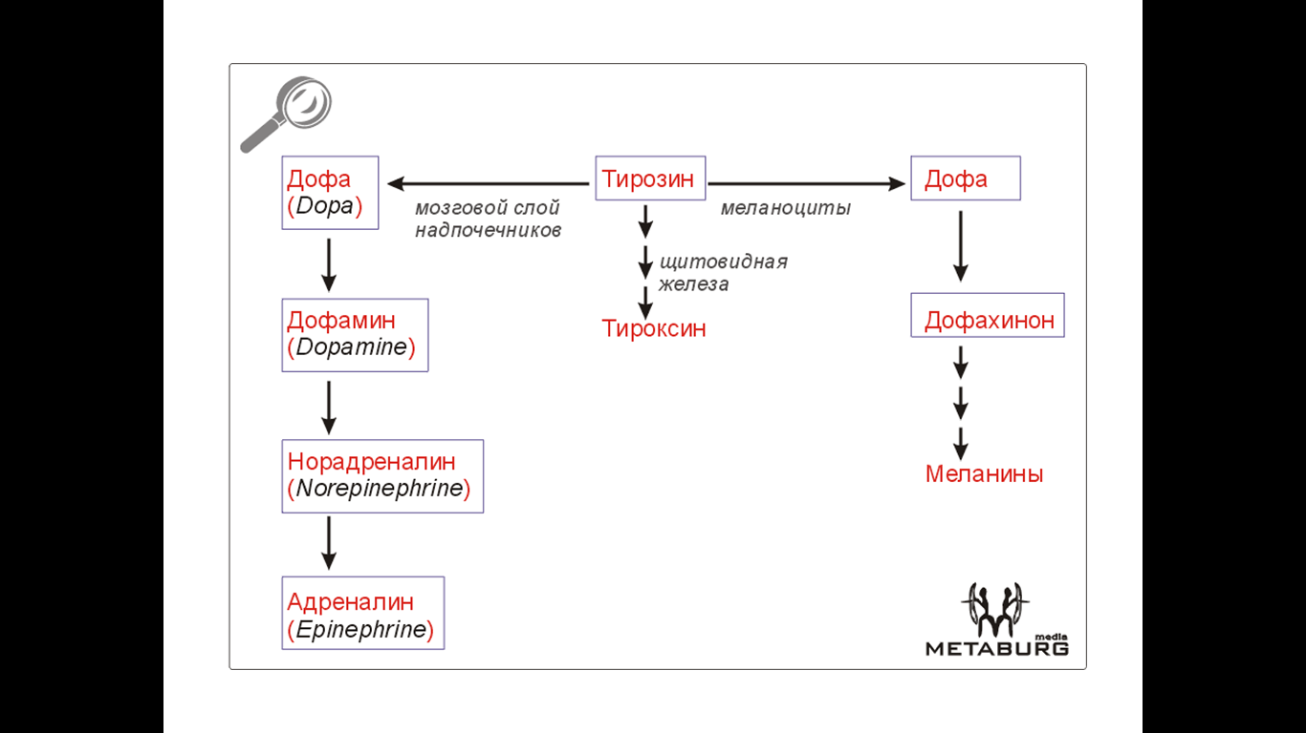
· Альбинизм. Причина - нарушение образования меланина из тирозина. Наиболее распространенная аутосомно-рецессивная форма (еще м.б. аутосомно-доминантная и сцепленная с полом) основывается на дефекте фермента меланобластов тирозиназы, в норме превращающая тирозин в 3,4-диоксифенилаланин.
Проявления – депигментация волос, сетчатки (вследствие чего родопсин распадается ускоренно => фотофобия), фотодерматит.
· Тирозиноз Медеса. Причина – нарушение активности оксифенилпируватдезоксигеназы. По другим данным, причина в дефиците митохондриальной печеночной тирозинаминотрансферазы. Гомогентизиновая кислота в печени вообще не образуется (в противоположность алкаптонурии).
Развиваются печеночная недостаточность, нефропатия.
· Гипертирозинемия 1 типа. Дефект фумарилацетоацетатгидролазы. Накапливаются тирозин и метионин.
Больные гибнут в младенчестве от печеночной недостаточности (цирроз печени) и нефропатии (тубулонекроз).
2 типа. Дефицит печеночной тирозинаминотрансферазы. Задержка психомоторного развития.
· Хоукинсурия. Дефект 4- гидроксифенилпируватдезоксигеназы. В отличие от остальных миноацидопатий, она аутосомно-доминантная. Тирозинемия.
Задержка психомоторного развития.
· Наследственный первичный гипотиреоз. Дефект йодтирозиндейодиназы. Моно- и дийодтирозин не дейодируются, развивается нехватка тиреоидных гормонов.
Зоб. Нарушение развития мозга и обучаемости.
· Паркинсонизм (см. схемку выше).
96. Нарушения композиции белков плазмы. Значение отдельных белковых фракций плазмы и различных плазменных белков при патологических процессах. Диспротеинемия, гипопротеинемия, гипоальбуминемия, их патологическое значение. Парапротеинемия, виды и этиология. Протеинурия, виды и патофизиологическое значение.
Метод электрофореза на бумаге, с подвижной границей выявляет в плазме здорового человека 5 основных фракций — альбуминовую (54-58%), а также 4 фракции глобулинов — α1 (6-7 %), α2 (8-9 %), β1 (13-14%) и γ (11-12%). Альбумин - основной компонент плазмы, обеспечивающий онкотическое давление (затем α1-глобулины). Факторы комплемента: от α2-фракции (С1s, C9) до γ-глобулиновой (Clq, C8); большинство β1 (СЗ-С5) и β2 (С2, С6, С7). Факторы свёртывания и противосвёртывающей системы в β1-β2-глобулиновой фракции.
Нормальное количество и фракционное распределение - эупротеинемия. Нарушения протеинограммы плазмы – диспротеинемии: ↑ концентраций (гиперпротеинемии), их ↓ (гипопротеинемии) и появление в плазме необычных белков, в норме не присутствующих в значимых количествах (парапротеинемии).
Ложная гиперпротеинемия - при сгущении крови (гиперосмолярной дегидратации). Истинная только при парапротеинемиях, это почти всегда гиперглобулинемия.
Псевдогипопротеинемия - при гемодилюции. Истинные делят на первичные (наследственные) и вторичные (приобретенные). Первичная гипоальбуминемия - у недоношеных (незрелая печень). Наследственная анальбуминемия: альбумины менее 3% белка плазмы, компенсаторно увеличено абсолютное содержание глобулинов. Врождённая агаммаглобулинемия - при Х-сцепленном рецессивном синдроме Брутона (ниже 1 г/л). Гипогаммаглобулинемия - простой вариабельный иммунодефицит. Первичная гипопротеинемия с понижением и альбумина, и глобулинов, особенно γ, - при экссудативной энтеропатии у детей (потеря белка при диарее).
Наиболее важные этиологические факторы вторичных гипопротеинемий:
Пищевая белковая недостаточность (квашиоркор, алиментарный маразм).
Нарушения поступления АК из кишечника при адекватной диете (хронические и рецидивирующие энтериты: целиакия, инфекционные формы спру; наследственные дефекты переваривания и всасывания белков, панкреатическая недостаточность).
Печёночная недостаточность: тормозятся переаминирование и восстановительное аминирование, нарушается балансовая функция в отношении смеси пищевых аминокислот -> гипераминоацидемия, аминоацидурия и дефицит белков печёночного происхождения в плазме: альбумин, трансферрин, протромбин, фибриноген, факторы свёртывания V, VII, IX—XIII. Если болезнь, вызвавшая угнетение функций печени, имеет инфекционную или иммунопатологическую природу – гипербетагаммаглобулинемия (преиммунный и иммунный ответ).
Усиленные внекишечные потери белка через почки. Протеинурия ренальная, преренальная и постренальная. Почечные механизмы протеинурии: повышенная проницаемость клубочкового фильтра и возрастание фильтрации белка, превышающее максимальную производительность системы реабсорбции (острый диффузный гломерулонефрит), понижение реабсорбции белка при неизменной фильтрации (первичный нефротический синдром), увеличение выделения белка эпителием канальцев (синдром Фанкони). При иммунопатологиях – комбинация механизмов. При изолированном нарушении реабсорбции степень селективности высока (наиболее мелкодисперсный белок — альбумин). Если протеинурия неселективна, альбумин в моче до 50%, есть все основные фракции плазменных белков и обычно не менее 10 % - γ-глобулины. Преренальные причины: парапротеинемии. Постренальные причины: гиперсекреция желез мочевыводящих путей и протеинурия, смешанная с пиу- и гематурией (инфекции урогенитального тракта).
Некишечные потери через кожу — плазморея при ожоговой болезни (потеря IgG), эксфолиативный дерматит и синдромы Лайелла и Стивенса-Джонсона + при ↑ гнойных экссудатах (эмпиема), но патогенез затяжных гнойных процессов предусматривает системное действие кахексина и др. цитокинов, вызывающих острофазный ответ, и здесь ограничение синтеза альбумина важнее прямых потерь белка.
Резко увеличенная проницаемость капилляров и венул при шоке, системном действии медиаторов воспаления, системных васкулитах - потеря белков плазмы путём перераспределения между кровью и тканями при недостаточном возврате белка через лимфатические пути. Лимфодинамический отёк при филяриозах («элефантиаз»), конечности не отдают избыток белка из интерстиция в силу окклюзии лимфатических сосудов -> могут превратиться в депо белка, а в крови возможна гипопротеинемия.
При преиммунном ответе TNF, IL-1, 6, 8 вызывают ↑ синтеза гепатоцитами и МФ положительных глобулинов острой фазы, ↓ производства альбумина и трансферрина (отрицательного глобулина острой фазы). Биологический смысл в повышении антиокислительной резистентности тканей, ограничении масштабов альтерации, индукции гипоферремии и гипоцинкемии, что ↓скорость размножения многих бактерий.
Этиология парапротеинемий связана с неопластическими клональными пролиферациями мутантных линий плазмоцитов -> синтез Ig или их отдельных цепей.. При миеломной болезни (Рустицкого-Калера) множественные очаги — колонии миеломных клеток в костях, могут вырабатывать IgG, IgM, IgD, IgA, IgE или свободные лёгкие, либо тяжёлые цепи Ig. Особая форма (только тяжёлые α-цепи Ig) - иммунопролиферативная энтеропатия (средиземноморская лимфома), поражает лимфоидную ткань тонкого кишечника. При плазмоцитоидной лимфоцитарной лимфоме (макроглобулинемия Вальденстрёма) ↑IgM. Ig легко агрегируют на холоде -> холодовой феномен Рейно. При всех парапротеинемиях в моче легкие цепи Ig — белки Бенс-Джонса. Менее значительно количество Ig может возрастать при действии поликлональных иммуностимуляторов.
Криоглобулинемии - нарушения композиции глобулинов плазмы, при которых она имеет тенденцию к формированию желе на холоде (из парапротеинемий встречается только при IgM-секретирующих миеломах и лимфомах).
При патологии из-за деструкции клеток в плазме могут оказываться повышенные количества различных ферментов -> всегда маркёр цитолиза.
97. Нарушения белкового обмена при эндокринных заболеваниях. Патология конечных этапов обмена белка. Гиперазотемия. Креатинурия. Уремия. Гипераммониемия. Этиология, патогенез, виды. Последствия. Роль при возникновении различных видов комы.
Нарушения белкового обмена. Усиление роста свидетельствует об активации синтеза белков или торможении их разрушения. Действительно, введение СТГ животным вызывает задержку азота в организме, положительный азотистый баланс и понижение распада белков. При этом установлено увеличение включения разных аминокислот в белки тканей и снижение отношения остаточного азота к белковому.
Считается, что действие СТГ опосредовано действием пептидных ростовых факторов - инсулиноподобных факторов роста (ИФР), синтезируемых в тканях и прежде всего в печени. Именно с их действием связывают такие анаболические эффекты, как:
1) стимуляция включения SO4 в протеогликаны;
2) стимуляция включения тимидина в ДНК;
3) стимуляция синтеза РНК;
4) стимуляция синтеза белка СТГ. Анаболический эффект СТГ обусловливают два момента:
1. Наличие инсулина. На фоне экспериментального диабета у животных и сахарного диабета у людей СТГ обычно не усиливает синтеза белков. Очевидно, это связано с тем, что инсулин активирует обмен углеводов и стимулирует синтез белка.
2. Концентрация глюкокортикоидов. Малые их дозы способствуют реализации анаболического эффекта СТГ, а большие дозы, наоборот, тормозят анаболический эффект СТГ и задерживают рост, что может быть связано с тем, что кортизол в больших дозах угнетает образование ИФР. У больных с эозинофильной аденомой гипофиза часто усилена продукция глюкокортикоидов. Не исключено, что это один из компенсаторных процессов, направленных на ограничение эффекта избыточных количеств СТГ.
Нарушения белкового обмена при отсутствии инсулина характеризуются преобладанием процессов катаболизма вследствие активации глюконеогенеза из глюкогенных аминокислот и снижения проницаемости клеточных мембран для аминокислот, что приводит к недостатку в тканях свободных аминокислот и нарушению процесса синтеза белка. Стимулируется синтез мочевины, что характеризуется гиперазотемией и приводит к отрицательному азотистому балансу.
Диабетическая кома. Критическая дегидратация тканей организма с поражением
функций головного мозга ведет к развитию диабетической (гипергликемической)
комы. Кома развивается при достижении концентрации глюкозы в крови от 19,4 до 33,3 ммоль/л и более. В этих условиях вследствие кетоацидоза ионы калия выходят во внеклеточное пространство (гиперкалиемия), что лежит в основе нарушения
сократительной функции миокарда, а также дыхательной мускулатуры. Диабетическая кома может привести к летальному исходу, если больному не будет своевременно проведена специфическая противокоматозная терапия.
Различают следующие виды диабетической комы:
1. Гипергликемическая кетоацидотическая кома. Развивается чаще всего у больных СД 1 типа вследствие гипергликемии, гиперкетонемии и метаболического ацидоза.
Глюкоза и кетоновые тела выводятся с мочой (глюкозурия и кетонурия), что способствует увеличению осмотического давления в первичной моче, потере ионов Na и сопровождается полиурией. Содержание глюкозы в крови превышает 22 ммоль/л, кетоновых тел - 17 ммоль/л, повышено содержание остаточного азота, мочевины, холестерина, жирных кислот, уровень натрия чаще нормальный, реже - снижен, уровень калия чаще нормальный, у больных с почечной недостаточностью может быть повышен.
2. Гипергликемическая гиперосмолярная кома. Встречается реже, чем кетоацидотическая, и развивается у больных СД 2 типа старше 50 лет при дополнительном воздействии обезвоживающих факторов (рвота, понос, ограничение приема жидкости, ожоги, кровопотеря, полиурия, прием диуретиков). Основными звеньями патогенеза этого вида комы являются дегидратация организма и развитие гиперосмолярности плазмы, уровень гликемии может достигать 55 ммоль/л. У больных нет выраженной гиперкетонемии и кетонурии, отсутствует запах ацетона изо рта и, если не обратиться к врачу, нарастает уровень глюкозы в крови до крайне высокой степени, что способствует усилению диуреза (глюкозурический осмотический диурез). Возникающее обезвоживание приводит к гиповолемии, стимуляции секреции альдостерона и задержке ионов Na и Cl. Показатель осмолярности плазмы повышается в 1,5-2 раза (в норме около 300 мосмоль/л, при коме достигает 500 мосмоль/л), что приводит к резко выраженной
внутриклеточной дегидратации, нарушению водного и электролитного равновесия в клетках мозга, гипоксии ЦНС с выраженной неврологической симптоматикой и потере сознания.
3. Гипергликемическая кома с лактат-ацидозом (лактацидотическая). Это
относительно редкое, но опасное осложнение СД. В механизме ее развития важную роль играют следующие факторы:
а) снижение активности ферментативного пируватдегидрогеназного комплекса
(выявляется при дефиците инсулина), превращающего пируват в ацетил-КоА. Пируват в обратимой реакции, катализируемой лактатдегдрогеназой, превращается в молочную кислот
б) применение лекарственных препаратов, стимулирующих анаэробный гликолиз и тем самым повышающих содержание лактата и пирувата в организме (например, бигуаниды, повышающие утилизацию глюкозы за счет ее анаэробного распада). При поражении печени или почек может иметь место кумуляция этих препаратов в организме, в результате чего развиваются лактоацидоз и кома;
в) гипоксическое состояние (при котором, как правило, стимулируется гликолиз),
вызванное физическим переутомлением, сердечной или дыхательной недостаточностью.
Гиперкетонемия и кетонурия отсутствуют, могут выявляться незначительная гипергликемия и небольшая глюкозурия. Вследствие несвоевременной диагностики и трудности лечения прогноз может быть неблагоприятным.
4. Гипогликемическая кома. Связана с передозировкой инсулина, препаратов сульфонилмочевины, развитием вторичного гипопитуитаризма (следствие ангиопатии сосудов гипофиза), ослабляющего ответ на гипогликемию, и явлениями диабетического нефросклероза, что удлиняет время циркуляции инсулина и, кроме того, еще более снижает почечный порог для глюкозы, способствуя ее потере.
Причинами гипогликемии могут быть также гиперпродукция инсулина опухолью
поджелудочной железы (инсулиномой), недостаточность контринсулярных гормонов, печеночные формы гликогенозов, заболевания печени, голодание, нарушение расщепления и всасывания углеводов в желудочно-кишечном тракте и др.
В механизме развития гипогликемической комы решающее значение имеет снижение доставки глюкозы к нервным клеткам, что ведет к их энергетическому истощению и нарушению функций ЦНС. При снижении уровня глюкозы менее 3 ммоль/л возникают потливость, тремор, чувство тревоги и голода, слабость. Затем развивается состояние, напоминающее алкогольное опьянение и сопровождающееся дезориентацией, агрессивностью, галлюцинациями. При дальнейшем падении содержания глюкозы (менее 2,5 ммоль/л) возникают клонические судороги и потеря сознания. В тяжелых случаях могут наступать отек и некроз отдельных участков мозга.
Креатинурия в норме характерна только для новорожденных и беременных женщин.
Увеличение выведения креатина с мочой наблюдается
· при мышечной атрофии (наследственные миодистрофии, генерализованной миастении, миозитах)
· голодание
· гипертиреоз
· любые пролонгированные состояния с отрицательным азотистым балансом
Повышение выведения креатина сопровождается уменьшением экскреции креатина и потерю белка.
Уремия -синдром аутоинтоксикации, развивающийся при выраженной почечной недостаточности, в результате задержки в организме азотистых метаболитов и других токсических веществ, расстройства водно-солевого, кислотно-щелочного и осмотического гомеостаза, сопровождающийся вторичными обменными и гормональными нарушениями, общей дистрофией тканей и дисфункцией всех органов и систем.
Ведущую роль в патогенезе уремии, как острой, так и хронической, играет интоксикация продуктами обмена, в норме выводящимися с мочой. Доказано, что в организме больных с уремией аккумулируется большое число органических веществ, особенно продуктов белкового метаболизма, многие из которых обладают токсичностью. Кроме мочевины накапливаются, в частности, аммиак, цианат, креатинин, гуанидины, мочевая кислота, β2-микроглобулин, β2-глюкопротеин, пептиды средней молекулярной массы, аминокислоты, дериваты пиридина, алифатические и ароматические амины, полиамины, индол, фенолы, миоинозитол, маннитол, ацетон, липохромы, циклический АМФ, глюкуроновая и щавелевая кислоты, ряд гормонов, некоторые ферменты и другие.
При ХПН гиперазотемия обусловлена в основном повышением содержания мочевины (ее доля составляет до 80 % от всего небелкового азота), с накоплением которой, помимо диуретического эффекта, связывают появление у больных апатии, тошноты, головной боли и других клинических симптомов. Различная реакция больных на одну и ту же степень гиперазотемии, очевидно, обусловлена двумя факторами: 1) индивидуальной чувствительностью к гиперазотемии. Отдельные больные отмечают плохое самочувствие при умеренной гиперазотемии (концентрация мочевины — 10— 13 ммоль/л). Вместе с тем мы наблюдали девочку 14,5 лет с ХПН вследствие нефронофтиза Фанкони, которая посещала школу при повышении концентрации сывороточной мочевины до 43 ммоль/л; 2) степенью нарушения в этот период других гомеостатических функций почек (ацидоз, водно-электролитные расстройства и др.).
Гипераммониемия — это нарушение обмена веществ, проявляющееся в недостаточности цикла ферментов мочевины, приводящее к отравлению организма аммиаком. Симптомы аммиачного отравления проявляются при превышении этих пределов всего в 2—3 раза. Предельно допустимый уровень аммиака в крови 60 мкмоль/л. При повышении концентрации аммиака (гипераммониемия) до предельных величин может наступить кома и смерть. При хронической гипераммониемии развивается умственная отсталость.
Транзиторной гипераммониемией называется также пограничное состояние, присущее новорожденным детям в период адаптации к внеутробной жизни, проявляющееся обычно на вторые—третьи сутки жизни. Этот вид гипераммониемии встречается чаще всего у недоношенных детей с задержкой внутриутробного развития, с частотой до пятидесяти процентов рождений, однако иногда регистрируется и у доношенных малышей. Часть детей не проявляет симптоматики клинической картины гипераммониемии: признаки угнетения ЦНС (вялость, понижение мышечного тонуса, приступы апноэ, ослабленная реакция зрачков на свет, отказ от еды, ступор и кома), а также расстройства дыхательной функции, желтуха, судороги и обезвоживание. Причиной, вызывающей гипераммониемию, называют кислородное голодание, или гипоксию, во время беременности и в процессе родов.
Дата добавления: 2016-01-04; просмотров: 30; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!