Приобретённые формы
Приобретённая (вторичная) гипераммониемия развивается вследствие заболеваний печени и вирусных инфекций. В крайне тяжёлых случаях она проявляется как тошнота, рвота, судороги, нечленораздельная речь, затуманивание зрения, тремор, нарушение координации движений.
Наследственные формы гипераммониемии вызваны генетическим дефектом любого из пяти ферментов синтеза мочевины. Соответственно ферменту заболевание делится на пять типов. Первичными признаками гипераммониемий являются сонливость, отказ от пищи, рвота, беспокойство, судороги, нарушение координации движений, тахипноэ, дыхательный алкалоз. Могут развиться печёночная недостаточность, лёгочные и внутричерепные кровоизлияния.
Наиболее частой является гипераммониемия типа II, связанная с недостатком орнитин-карбамоилтрансферазы. Заболевание рецессивно, сцеплено с Х-хромосомой. У матери также наблюдается гипераммониемия и отвращение к белковым продуктам. При полном дефекте фермента наследственные гипераммониемии имеют раннее начало (в период до 48 часов после рождения).
Лабораторным критерием заболевания является накопление глутамина (в 20 и более раз) и аммиака в крови, ликворе и моче.
Причины Гипераммониемии:
Токсичность аммиака обусловлена следующими обстоятельствами:
1. Связывание аммиака при синтезе глутамата вызывает отток α-кетоглутарата из цикла трикарбоновых кислот, при этом понижается образование энергии АТФ и ухудшается деятельность клеток.
|
|
|
2. Ионы аммония NH4+ вызывают защелачивание плазмы крови. При этом повышается сродство гемоглобина к кислороду (эффект Бора), гемоглобин не отдает кислород в капиллярах, в результате наступает гипоксия клеток.
3. Накопление свободного иона NH4+ в цитозоле влияет на мембранный потенциал и работу внутриклеточных ферментов — он конкурирует с ионными насосами для Na+ и K+.
4. Продукт связывания аммиака с глутаминовой кислотой — глутамин — является осмотически активным веществом. Это приводит к задержке воды в клетках и их набуханию, что вызывает отёк тканей. В случае нервной ткани это может вызвать отёк мозга, кому и смерть.
98. Диспротеинозы. Амилоидоз, этиология, патогенез, виды. Роль при патологии. Происхождение и свойства амилоида. Понятие о гиалинозе и роговой дистрофии. Механизмы их формирования.
Диспротеинозы (белковые дистрофии) – процессы накопления в клетках и межклеточном веществе количественно и качественно измененных продуктов обмена белков.
В рез-те гипоксии возникают: мутное набухание, гидропическая дистрофия; вследствие альтерации межклеточного вещества при воспалении и иммунопатологических процессах – мукоидное и фибриноидное набухание). Механизмы мутного набухания (зернистой дистрофии) трактуются как корреляты ранней стадии обратимой гипоксии клетки; механизмы гидропической – как корреляты более глубокой клеточной гипоксии. Мукоидное и фибриноидное набухание – как начальные и глубокие, сопровождаемые коагуляцией, стадии альтерации межклеточного вещества при воспалении. Гиалиново-капельная дистрофия связана с патологией цитоскелета и агрегацией промежуточных филаментов. Также возможно накопление липопротеидов, гликопротеидов, производных аминокислот и т.д.
|
|
|
Амилоидоз – патофизиологический процесс, относительно которого есть явные свидетельства, что в большинстве случаев он связан с определенными расстройствами в иммунном аппарате. Амилоидоз следует рассматривать как группу нарушений, объединяемых по принципу отложения сходно устроенных белковых комплексов. Амилоид – патологический белковый комплекс, который на окрашенным гематоксилином и эозином препаратах выглядит как розовый прозрачный материал, депонированный между клетками в различных тканях и органах. Он обнаруживается при большом числе разнообразных синдромов и болезней.
|
|
|
Все типы амилоида обладают следующими свойствами:
· Окрашивание в красный цвет при окраске йода с последующим изменением цвета на синий или фиолетовый после обработки разбавленной серной кислотой;
· При окраске гематоксилином и эозином в световом микроскопе амилоид выглядит как аморфная, эозинофильная, розовая, прозрачная, внеклеточная субстанция;
· При окраске метиловым фиолетовым – метахромазия;
· Амилоид метится различными антисыворотками против его компонентов при иммуноморфологическом анализе и дает люминесценцию с рибофлавинами S и T.
Различают системный амилоидоз (первичный-связан с иммунодискразиями и вторичный-осложнение основных заболеваний) и местный. Отдельно выделяют наследственный амилоидоз. При первичном наиболее часто вовлекаются почки, средце, печень, селезенка, лимфоузлы, суставы, лучезапястные суставы, кожа, нервы, язык. Вторичный может вовлекать печень, селезенку, почки, сердце, надпочечники.
Имеются два главных элемента амилоидных фибрилл: 90% - фибриллярный компонент, 10% - Р-компонент. Фибриллы белка амилоида образуют типичную уникальную структуру – бета-складчатую листоподобную структуру, не встречающуюся у млекопитающих. Р-компонент, выглядящий как палочка при электронной микроскопии, на разрезе – пятиугольник, составленный из пяти шарообразных структур.
|
|
|
Два типа фибриллярного компонента: AL и AA. Первый формируется при участии плазматических клеток и обычно содержит легкие цепи Ig или иногда их N-концевые участки. Миеломная болезнь (моноклональная злокачественная опухоль), В-клеточные лимфомы осложняются амилоидозом, т.к. ненормальные плазмоциты производят составные части амилоида AL. При образовании злокачественного клона В-клеток происходит то же
самое; причем чем злокачественнее клон, тем меньше синтез амилоида и наоборот. Амилоидоз типа AL, произведенным В-клетками, называется первичным (иммунопатологическое расстройство).
Тип АА – этот белок является белком неиммуноглобулиновой природы, предшественники которого синтезируются в печени и, по-видимому, макрофагами. Белок не имеет структурной гомологии с какими-либо белками, сод-ит 76 АК, хар-н для вторичного амилоидоза. Амилоидный предшественник в сыворотке обозначается SAA, циркулирует в составе альфа1-глобулинов в комплексе с ЛПВП третьего подкласса. SAA имеет три формы и принадлежит к положительным глобулинам острофазного ответа. В амилоидогенезе участвуют продукты неполного протеолиза SAA, создаваемые макрофагами, которые захватывают дериваты этого белка в составе иммунных комплексов. Соответственно для обоих типов амилоидоза хар-на связь с длительной активацией иммунной системы и необходима обработка и сборка их компонентов в активных макрофагах.
Кроме предыдущих двух были найдены другие белки в амилоидах:
· Транстиретин (преальбумин) – нормальный сывороточный белок, транспортирующий тироксин и ретинол. Мутантная его форма депонируется при семейной амилоидной нейропатии и семейной амилоидной миокардиодистрофии – аутосомно-доминантные расстройства. Может образовываться без участия фагоцитов.
· Белок бета2-микроглобулин – найден в составе амилоида у пациентов после длительного диализа.
· Бета2-белок амилоида – обнаружен при болезни Альцгеймера. Иногда данный амилоидоз называют старческим мозговым.
Р-компонент идентичен сывороточному альфа1-гликопротеиду, высокогомологичен С-реактивному белку; по-видимому, является острофазным белкоми его синтез усиливается при преиммунном ответе. Обеспечивает положительную реакцию с реактивом Шиффа.
Гиалиноз – накопление в соединительной ткани (включая строму и стенки сосудов) любого неамилоидного белкового, гомогенного, плотного, эозинофильного материала. Т.о., одни и те же иммунопатологические процессы могут способствовать депонированию и амилоидного, и неамилоидного белка. Основные механизмы образования: плазматическое пропитывание (гиалин содержит белки плазмы и иммунные комплексы), местная денатурация и коагуляция белков при воспалении и иммунопат. процессах с аутоАТ. Внутриклеточный гиалин при гиалиново-капельной дистрофии и внеклеточный различны. Например, тельца Маллори – гиалиновые включения в гепатоцитах, образовавшиеся в связи с агрегацией белка прекератина, входящего в состав промежуточных филаментов цитоскелета. Тельца Русселя – агрегаты фрагментов АТ в плазм. клетках при интенсивном синтезе Ig. => внутриклеточный гиалин различается по механизму образования.
Роговая дистрофия – избыточное образование кератина при ускорении клеточного цикла и апоптотической гибели кератиноцитов эпидермиса, что может приводить к гиперкератозу и дискератозу (наследственный ихтиоз). Ей способствует гиповитаминоз А. Гиперкератоз характерен также для доброк. опухолей из базальных клеток эпидермиса; образ-ние «раковых жемчужин» - лейкоплакия в карциномах.
99. Нарушения обмена нуклеопротеидов. Причины и механизм нарушений. Гиперурикемия и ее патогенные последствия. Этиология и патогенез подагры. Нарушения обмена пиримидиновых нуклеотидов.
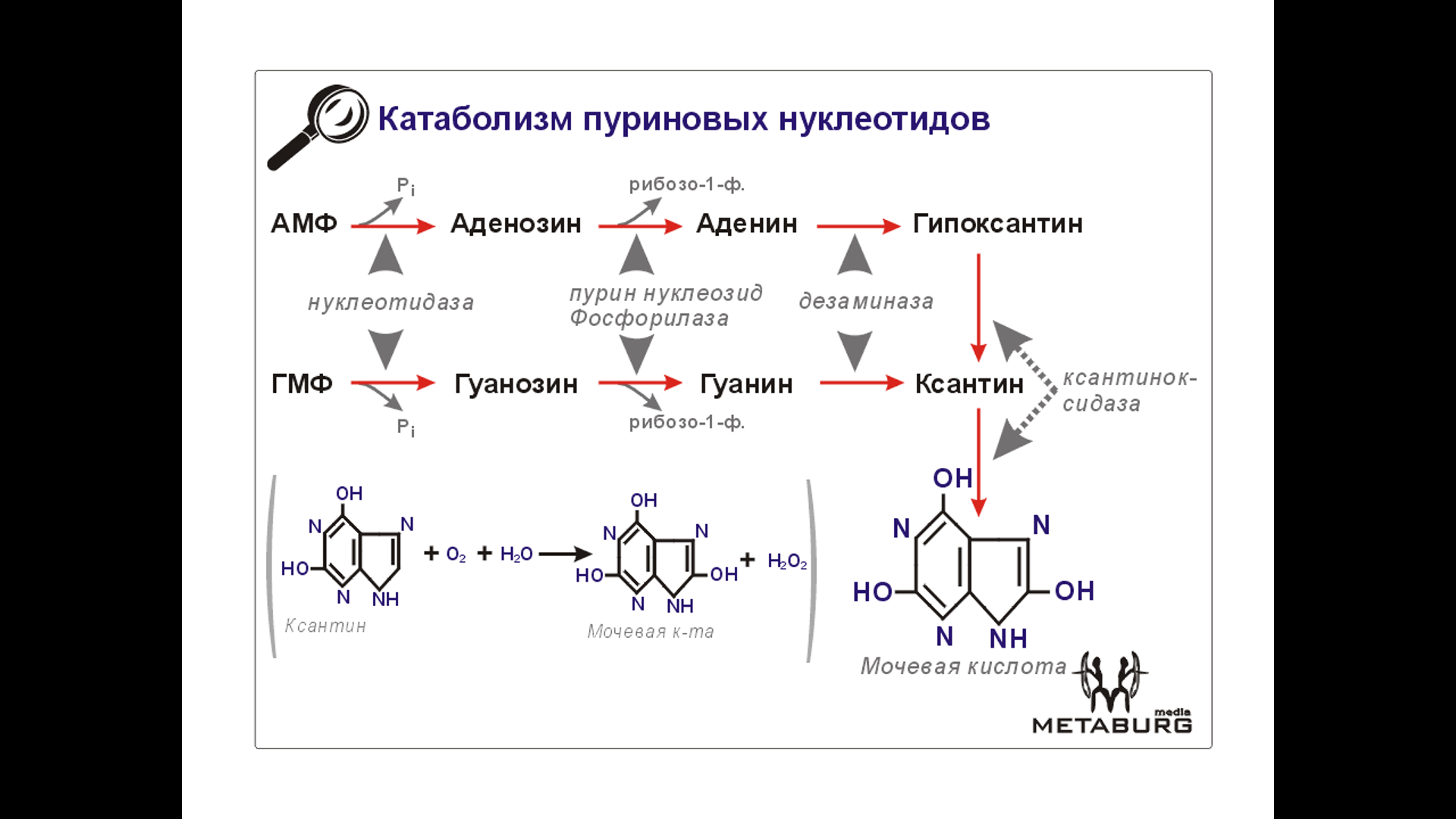
При нарушении катаболических процессов в крови происходит повышение концентрации мочевой к-ты – гиперурикемия.
Повышение уровня мочевой кислоты увеличивает предрасположенность к подагре и почечной недостаточности.
Этиология
Причины первичной гиперурикемии (идиопатическая или врожденная):
1. Около 1% людей, имеющих гиперурикемию, имеют генетический ферментный дефект метаболизма пуринов, который приводит к перепроизводству мочевой кислоты
2. Нарушение различных фаз выведения мочевой кислоты
Причины вторичной гиперурикемии:
1. повышенное образование мочевой кислоты
2. замедление выведения мочевой кислоты
Мочевая кислота — слабокислый продукт, поэтому она, секретируясь в кровь, образует соли-ураты — со щелочными катионами (на 98 % — с натрием), которые связываются глобулинами плазмы только на 5 % и выводятся на 2/3 почками, а на 1/3 — через тонкий кишечник.
Ураты легче выпадают в осадок при температурах ниже 37 °С. Наиболее «холодным» суставом организма является первый предплюснофаланговый сустав стопы.
В моче кристаллизации уратов способствует кислый рН. Именно поэтому все виды ацидоза способствуют обострениям подагры.
Подагра в типичных случаях дебютирует после 40 лет и проходит 4 стадии.
Латентная гиперурикемическая стадия протекает без клинических симптомов и проявляется лишь выявляемой лабораторным анализом гиперурикемией.
Дебютная стадия представляет собой острый моноартрит.
Моноартрит начинается внезапно, на фоне предшествующей гиперурикемии и повышенного содержания уратов в синовиальной жидкости. Подагрический моноартрит характеризуется острейшими признаками воспаления — отеком, краснотой, ограничением функции, повышением местной температуры, а часто — и общей лихорадкой.
Главным механизмом острого артрита служит реакция сторожевой полисистемы плазмы и полиморфонуклеарных лейкоцитов, в первую очередь — нейтрофильных, на полианионные кристаллы уратов.
1) Ураты активируют классический и альтернативный пути комплемента, фактор Хагемана, а через них — и всю контактную систему полипептидных медиаторов, включая кинины, свёртывание, фибринолиз.
2) Взаимодействие уратовых кристаллов с полиморфонуклеарами и фагоцитами, приводит к освобождению и активации воспалительных медиаторов.
3) Гибель нейтрофилов при фагоцитозе кристаллов ведет к освобождению активных кислородных радикалов, а также особого кристаллозависимого хемотактического фактора, дефензинов и огромного количества лизосомальных гидролаз. Кристаллозависимый хемоаттрактант лейкоцитов, возможно, один из главнейших участников этого «пожара».
4) Макрофаги, фагоцитируя ураты, активируются и выделяют цитокины ИЛ-1, ИЛ-6, ИЛ-8, кахексии, а также простагландины. Это усиливает воспаление и приводит к выделению синовиоцитами коллагеназ, поддерживающих альтерацию.
Самоограничение симптомов артрита зависит от выработки противовоспалительных медиаторов, главным образом, макрофагального и синовиоцитарного происхождения.
Межприсгпупный период подагры характеризуется отсутствием симптомов острого артрита. Несмотря на это сохраняется гиперурикемия, происходит отложение уратов в тканях, прогрессирует нефропатия, и бывают повторные атаки артрита, которые вовлекают тот же и новые суставы. Как и первый приступ, повторные возникают в ответ на тот же разнообразный спектр провоцирующих агентов.
Период хронического продуктивного артрита
С течением времени (как правило, по прошествии 8-10 лет от первого приступа) в пораженных суставах и вокруг них образуются в результате гранулёматозного воспаления подагрические шишки —тофусы. Это деформирует суставы. Кожа над ними может изъязвляться, имеется персистирующий полиартрит с умеренным болевым синдромом, хотя сами шишки на ощупь вне приступа парадоксально безболезненны.
Главные механизмы этого периода подагры связаны с хронической активацией макрофагов, внутри которых персистируют фагоцитированные кристаллы. Это ведет к накоплению цитокинов, формирующих своего рода подагрическую гранулёму — очаг хронического пролиферативного воспаления.
Изменения в почках
При подагре накопление мочевой кислоты и уратов ведет, вследствие кристаллизации, к образованию камней в почках. Уролитиазу способствуют кислая реакция мочи и инфекция. Характерно, что сами камни состоят, главным образом, из мочевой кислоты, в чистом виде или вместе с оксалатом кальция (84%). Урат натрия не участвует в камнеобразовании. Мочевая кислота вступает в перекристаллизацию совместно с оксалатами. Пуриновая нагрузка провоцирует оксалурию.
Сами кристаллы мочевой кислоты и оксалатов могут закупоривать канальцы.
Взаимодействие уратов с медиаторами воспаления и лейкоцитами в почке, как и в суставе, ведет к воспалению. Формируется интерстициальныи и пирамидальный нефрит. Как и в суставах, в почке могут возникать гранулёматозные поражения и даже подагрические шишки. Присоединение инфекционного компонента способствует развитию хронического пиелонефрита.
Клинически, течение нефропатии может быть латентным, но могут отмечаться протеинурия и гипертензия. Исходом являются нефросклероз и хроническая почечная недостаточность, причем по мере ее формирования уровень гиперурикемии растет и артрит отягощается. Без лечения 25-30 % больных подагрой умирают от уремии.
Дата добавления: 2016-01-04; просмотров: 39; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!