Основные показатели КОС крови
В клинической практике для оценки кислотно-основного состояния используют следующие показатели:
рН - величина отрицательного десятичного логарифма молярной концентрации ионов водорода.крови рН артериальной крови (плазмы) при 37°С колеблется в пределах 7,40+-0,04 (слабощелочная реакция)
рН плазмы крови можно определить двумя методами:
1. Индикаторный метод основан на свойстве некоторых слабых кислот или оснований, используемых в качестве индикаторов, дисоциировать при определенных значениях рН и изменять свой цвет.
2. Метод рН-метрии позволяет более точно и быстро определять концентрацию протонов с помощью полярографических электродов, на поверхности которых при погружении в раствор создается разность потенциалов, величина которой зависит от рН среды
Активный электрод, выполненный из золота или платины, является измеряющим, другой, референтный, служит электродом сравнения. Активный электрод отделен от остальной системы стеклянной мембраной, проницаемой только для протонов. Электроды погружают в исследуемый раствор (кровь) и поляризуют от источника тока. Поскольку активный электрод дополнительно отделен от раствора электролита стеклянной мембраной, проницаемой только от ионов водорода, величина напряжения на обеих поверхностях этой мембраны пропорциональна рН крови.

Рис.9. Схема полярографической регистрации рН раствора (рН-метрии).
|
|
|
Реакция мочи, как правило, слабо кислая - рН мочи находится в пределах от 5 до 7. Почки играют ведущую роль в регуляции кислотно-основного состояния посредством выведения ежесуточно продуцируемых протонов. Несмотря на наличие буферной системы в почечных канальцах, позволяющей осуществлять максимальную экскрецию ионов водорода при минимальном снижении рН мочи, значение рН мочи обычно кислое (в среднем около 6). Значение рН мочи измеряют в течение 2 часов после взятия пробы, поскольку она становится более щелочной по мере хранения.
рСО2 – парциальное давление углекислого газа в крови. Этот показатель отражает количество растворенного углекислого газа в плазме, что является одной из нескольких форм, в которых он присутствует в крови. Углекислый газ также входит состав угольной кислоты (Н2СО3), иона бикарбоната (НСО3-), в комбинации с белками плазмы и в связанном виде с гемоглобином (карбаминогемоглобин). Основное количество углекислого газа попадает в эритроциты по градиенту концентрации, где, за счет активности карбоангидразы превращается в угольную кислоту, после диссоциации которой протоны связываются гемоглобиновым буфером, а бикарбонат поступает в плазму в обмен на анионы хлора (хлоридный сдвиг). Процесс связывания ионов водорода усиливается по мере прохождения крови через капилляры и отдачи кислорода тканям, так как гемоглобиновый буфер обладает большей буферной емкостью в восстановленном состоянии.
|
|
|
В легких процесс идет в противоположном направлении из-за низкого парциального давления углекислого газа в альвеолярном воздухе, углекислый газ удаляется с выдыхаемым воздухом При нормальной вентиляции рСО2 в крови находится в диапазоне 40±5 мм рт.ст.(4,7-6,0 кПа).
Накопление или уменьшение количества СО2 существенно влияют на кислотно-основное состояние. Углекислый газ растворяется в плазме и вступает в химическую реакцию, в которой вода и СО2 образуют угольную кислоту, диссоциирующую на ион водорода и ион бикарбоната:

Н2СО3 является слабой кислотой, а НСО3ˉ достаточно активным анионом. Таким образом, в результате растворения углекислого газа в плазме меняется рН. Так как растворение СО2 сопровождается образованием угольной кислоты, то накопление углекислого газа при гиповентиляции приводит к респираторному ацидозу. При гипервентиляции углекислый газ избыточно удаляется из организма и пополняется из нестойкой угольной кислоты. Происходит смещение уравнения влево в сторону образования угольной кислоты, снижается концентрация протонов, увеличивается рН, развивается дыхательный алкалоз.
|
|
|
При оценке концентрации бикарбоната плазмы с помощью анализаторов регистрируют 2 показателя – истинный (актуальный) бикарбонат (АВ) и стандартный бикарбонат (SВ).
АВ (истинный бикарбонат плазмы) – 19-25 ммоль/л. Рассчитывается при рСО2, определенном у данного конкретного больного. Уравнение Гендерсона-Гассельбаха для концентрации ионов бикарбоната решается следующим образом:
lg[НСО3]=рН+lg(рСО2´0,0307)-6,105
Расчет на основе этого выражения был рекомендован Международной Федерацией Клинической Химии (IFCC) для использования в клинико-диагностических исследованиях. рН и рСО2 измеряются для воды плазмы, поэтому концентрация бикарбоната эквивалентна его активности в воде плазмы.
SВ – стандартный бикарбонат плазмы.Содержание бикарбоната у данного больного в стандартных условиях (рСО2=40 мм рт.ст., рО2 = 100%, t=37°C), в норме колеблется в пределах 20-26 ммоль/л.
ВВ – буферные основания плазмы (по международной номенклатуре Buffer Bases), сумма всех компонентов буферных систем (бикарбонатной, фосфатной, белковой, гемоглобиновой), в норме находится в пределах 44-52 ммоль/л. Значение этого параметра в незначительной степени зависит от парциального давления углекислого газа в крови. Сопряжение между бикарбонатным и небикарбонатным буферными компонентами определяет устойчивость ВВ при остром изменении рСО2. В то же время ВВ существенно зависит от тканевого метаболизма и частично от функции почек. По величине ВВ возможно судить о сдвигах КОС, связанных с увеличением или уменьшением содержания нелетучих кислот в крови. Нормальная величина ВВ обозначается как норма буферных оснований (NВВ) или должная концентрация буферных оснований.
|
|
|
ВЕ– сдвиг буферных оснований, показатель недостатка или избытка буферных оснований (Bases Excess). Определяется разницей между истинной и должной концентрациями буферных оснований:
ВЕ=ВВ – NВВ
В норме значение определяется в пределах ВЕ±2,3 ммоль/л. При патологическом увеличении содержания оснований ВЕ становится положительным, при снижении – отрицательным. В последнем случае целесообразно использовать термин «дефицит буферных оснований».
Параметр ВЕ имеет важное клинико-диагностическое значение. Он позволяет определить степень метаболических нарушений КОС, вычислить общий недостаток или избыток оснований (ОВЕ) организма с помощью формулы:
ОВЕ=0,3´масса тела´ВЕ
Формы нарушений КОС
Сдвиги КОС обусловлены изменением концентрации ионов водорода в организме. Недостаточность резервных возможностей вызовет ацидоз или алкалоз.
Ацидоз – изменение КОС, при котором в крови появляется абсолютный или относительный избыток кислот.
Алкалоз – изменение КОС, характеризующееся абсолютным или относительным увеличением оснований в крови.
Степень выраженности как ацидоза, так и алкалоза может быть различной и определяется величиной рН.
Таблица
Степени выраженности нарушений кислотно-основного состояния
| Ацидоз | Алкалоз | Степень компенсации |
| 7,40 – 7,36 | 7,40 – 7,44 | Компенсированный |
| 7,35 – 7,31 | 7,45 – 7,49 | Субкомпенсированный |
| ≤ 7,30 | ≥ 7,50 | Декомпенсированный |
Классификация ацидозов и алкалозов


Нарушения кислотно-основного равновесия
| Нарушение кислотно-основного равновесия | Первичное нарушение | Респираторная компенсация | Почечная компенсация |
| Метаболический ацидоз | Снижение бикарбонатов | Гипервентиляция до снижения РСО2 | При отсутствии заболевания почек – повышение экскреции ионов Н+ и повышение реабсорбции ионов НСО3ˉ |
| Метаболический алкалоз | Повышение бикарбонатов | Гиповентиляция до повышения РСО2 | При отсутствии заболевания почек –снижение экскреции ионов Н+ и снижение реабсорбции ионов НСО3ˉ |
| Респираторный ацидоз | Повышение РСО2 | Нет | Повышение экскреции ионов Н+ и повышение реабсорбции ионов НСО3ˉ |
| Респираторный алкалоз | Снижение РСО2 | Нет | Снижение экскреции ионов Н+ и снижение реабсорбции ионов НСО3ˉ |
АЦИДОЗ
НЕГАЗОВЫЙ АЦИДОЗ
Метаболический ацидоз
Основной причиной возникновения этого варианта ацидоза служит нарушение тканевого метаболизма с образованием избыточного количества нелетучих эндогенных кислот (лактоацидоз, кетоацидоз). При нарушениях доставки кислорода метаболизм клеток перестраивается на путь анаэробного гликолиза с внутриклеточным накоплением ионов водорода за счет образования органических и неорганических кислот. Метаболический ацидоз часто связан с первичным нарушением обмена веществ, например, при диабете, голодании, лихорадке, при гипоксии вследствие нарушений кровообращения.
Лактатный ацидоз развивается при избыточном образовании молочной кислоты или снижении ее выведения из крови. Наиболее частой причиной его является нарушение процессов анаэробного окисления при гипоксии тканей у больных, находящихся в послеоперационном периоде, панкреатите, лейкозах, анемиях. Ацидоз активирует клеточные катепсины, усиливается распад белков, увеличивается содержание свободных аминокислот в крови. Лактатный ацидоз при нарушениях периферического кровотока может быть осложнением кетоацидоза.
Лактат образуется при анаэробном метаболизме глюкозы. При соответствующих условиях практически все ткани могут продуцировать молочную кислоту. Чаще всего лактоацидоз бывает транзиторным при шоке, послеоперационной травме, при интенсивной физической нагрузке, во время которой имеет место дисбаланс между доставкой кислорода и потребностью в нем сокращающихся мышц. Печень и, в меньшей степени, почки утилизируют лактат, используя его в энергетическом обмене или превращая его обратно в глюкозу. В большинстве клинических случаев лактоацидоз связан с неадекватным обеспечением тканей кислородом, как при шоке или остановке сердца. При таких условиях повышена продукция молочной кислоты и уменьшен ее клиренс из-за плохой перфузии печени. У пациентов с декомпенсированным лактоацидозом при шоке с выраженной тканевой гипоксией отмечают высокий процент летальных исходов.
Лактоацидоз может быть связан с нарушениями, при которых отсутствует гипоксия тканей. Такие варианты ацидоза описаны у пациентов с лейкозами, лимфомами, лимфогранулематозом и другими злокачественными новообразованиями, некорригированным диабетом, тяжелой печеночной недостаточностью. Механизмы, вызывающие ацидоз при этих состояниях, недостаточно ясны. При новообразованиях может локально повышаться тканевой метаболизм с продукцией лактата или может возникать препятствие кровотоку к некарциноматозным клеткам. В большинстве случаев отмечается обширное опухолевое поражение печени. Возможно, именно нарушение функции печени обусловливает накопление лактата в крови. Лактацидоз также часто развивается при нераспознанной ишемии или инфаркте кишечника, а также у больных с сердечной недостаточностью, получающих сосудосуживающие средства.
Различные лекарственные средства, ингибирующие функции митохондрий, также могут вызывать угрожающий жизни лактат ацидоз. К препаратам, способным оказывать такой эффект, относятся бигуаниды, использующиеся для лечения сахарного диабета и противовирусные нуклеозидные аналоги, используемые для лечения синдрома приобретенного иммунодефицита (AIDS).
Лактацидоз может наблюдаться при нарушении доставки и утилизации кислорода тканями (шок, сердечная недостаточность, тяжелая анемия, дефекты митохондриальных ферментов или ингибирование их окисью углерода и цианидами), а также при избыточном образовании или недостаточной утилизации L-лактата (гликогенозы, эпилептические припадки, сахарный диабет, алкогольная интоксикация. Этанол вызывает легкое повышение молочной кислоты, но при алкогольной интоксикации клинически значимый лактат ацидоз не развивается, если нет другихосложнений, таких как печеночная недостаточность., печеночная недостаточность, злокачественные новообразования).
Лактацидоз может быть обусловлен накоплением в крови D-лактата (продукта метаболизма бактерий кишечника) при синдроме слепой петли и кишечной непроходимости. В этих случаях неабсорбированные углеводы попадают в толстую кишку, где он превращаются в D-молочную кислоту при чрезмерно быстром росте грам-положительных анаэробов. У таких пациентов периодически возникают эпизоды метаболического ацидоза, к которым приводит употребление пищи, богатой углеводами. Проявления включают спутанность сознания, церебральную атаксию, невнятность речи и провалы памяти. У них могут появляться симптомы интоксикации. Лечение включает использование антимикробных препаратов для снижения числа D-лактат продуцирующих микроорганизмов в кишке наряду с диетой, содержащей ограниченное количество углеводов.
Лактоацидоз также может быть связан с наследственными нарушениями метаболизма: дефицитом глюкозо-6-фосфатазы или другими врожденными заболеваниями с дефектом глюконеогенеза или окисления пирувата. Лактоацидоз может развиваться при ослаблении оксидативного метаболизма в митохондриях. Лактоацидоз описан при генетических митохондриальных нарушениях, при которых идет процесс накопления молочной кислоты. Одно из подобныхзаболеваний, именуемое MELAS, включает митохондриальную энцефалопатию (МЕ), лактат ацидоз (LA) и эпизоды подобные инсультам (S). Дети с этой патологией на протяжении первых лет жизни жалоб не предъявляют, но затем начинают проявляться нарушения моторного и конгитивного развития. Митохондриальные дефекты также приводят к замедлению роста, припадкам и повторным инсультам. При проведении терапии снижение уровня молочной кислоты в плазме у детей с тяжелым лактат ацидозом может приводить к выраженному клиническому улучшению.
При нормальной функции печени происходит самостоятельная компенсация лактатного ацидоза, так как 50% его утилизируется в печени.
Клиническая картина тяжелого лактацидоза характеризуется угнетением сознания (от оглушения до сопора), тахикардией, гипотензией, частым глубоким «ацидотическим» дыханием, нарушением периферического кровообращения, и, часто, олиго-или анурией. При ацидозе развивается резистентность к сосудосуживающему действию катехоламинов, что приводит к развитию стаза и нарушению микроциркуляции, усугубляющим гипоксию. Уменьшение рН менее 7,2 может привести к снижению сердечного выброса.
Под влиянием гипоксии и ацидоза повышается уровень кининов в плазме и внеклеточной жидкости, что вызывает вазодилатацию и резкое повышение проницаемости сосудистой стенки. Усиление катаболизма при ацидозе ведет к гипергидратации за счет увеличения продукции эндогенной воды. В связи с тем, в экскрецию протонов вовлекаются слизистые ЖКТ и дыхательных путей, то возможно развитие ацидотического ларинготрахеобронхита, бронхиолита и альвеолита, гастроэзофагита с продолжительным кашлем, рвотой, иногда с примесью крови.
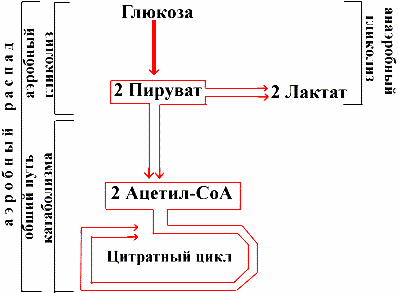
Рис.10. Схема лактоацидоза.
Кетоацидозразвивается при неполном окислении свободных жирных кислот, образующихся в избытке при усиленном липолизе и уменьшении образования в печени триглицеридов из-за низкой активности фермента ацилкарнитинтрансферазы.. Кетокислоты (ацетоуксусная и β-оксимасляная), образующиеся в печени из жирных кислот, являются источником энергии во многих тканях организма. Гиперпродукция кетокислот имеет место при недостаточном запасе углеводов или невозможности использования имеющихся доступных углеводов в энергетическом обмене. В этих условиях жирные кислоты мобилизуются из жировой ткани и поступают в печень, где превращаются в кетоны.Кетоацидоз развивается тогда, когда продукция кетонов превышает возможность их использования тканями.
Наиболее частая причина кетоацидоза – декомпенсированный сахарный диабет.
Диабетический кетоацидоз (ДКА) занимает первое место среди острых осложнений эндокринных заболеваний. Смертность при ДКА достигает 6-10%. При диабетическом кетоацидозе в крови накапливаются кетокислоты: ацетоацетат и β-гидроксибутират. ДКА возникает вследствие абсолютного или относительного дефицита инсулина, который может развиваться за несколько часов или дней.
У больных, получающих инъекции инсулина, причинами ДКА могут быть:
– назначение неадекватных (слишком низких) доз инсулина;
– нарушение режима инсулинотерапии;
– резкое возрастание потребности в инсулине: инфекционные заболевания (сепсис, в особенности уросепсис, пневмония., другие инфекции дыхательных и мочевых путей, менингит, синусит, холецистит, панкреатит и др.), сопутствующие эндокринные нарушения (тиреотоксикоз, синдром Иценко-Кушинга, акромегалия, феохромоцитома), инфаркт миокарда, инсульт, травмы, хирургические вмешательства, медикаментозная терапия (глюкокортикоиды, эстрогены, в том числе пероральные контрацептивы), беременность, стрессы (во всех этих случаях увеличение потребности в инсулине обусловлено увеличением концентрации контринсулярных гормонов – адреналина, кортизола, глюкагона и СТГ, а также резистентностью тканей к инсулину).
Вследствие нарастающего дефицита инсулина в организме происходит обеднение печени гликогеном, увеличение гликонеогенеза, и понижение утилизации глюкозы тканями. Нарастанию дефицита инсулина в немалой степени способствует и одновременно происходящее уменьшение количества рецепторов и их чувствительности к инсулину и в периферических тканях. Это приводит к значительной гипергликемии и глюкозурии. Развитие гипергликемии обусловлено не только дефицитом инсулина в организме, но и избыточной секрецией глюкагона, стимулирующего гликонеогенез и гликогенолиз. Вследствие гипергликемии увеличивается осмотическое давление во внеклеточной жидкости и развивается процесс клеточной дегидратации. Уменьшение содержания гликогена в печени обусловливает усиление мобилизации жира из депо с последующим поступлением его в печень. Это в конечном итоге приводит к жировой инфильтрации печени, а в последующем – к кетозу, который усугубляет дефицит инсулина. Следствием гиперкетонемии и кетонурии является нарушение водно-солевого обмена – понижение содержания натрия, фосфора, кальция, калия, магния и хлоридов в крови. Уровень калия в крови вначале повышен, а затем понижен, что связано с усиленным выведением его с мочой. Иногда возможна ранняя гипокалиемия, которая обусловлена массивной деструкцией клеток, потерей ими калия и интенсивной экскрецией его с мочой. Это, в свою очередь, ведет к дегидратации и сдвигу КОС в сторону ацидоза

Рис.11. Механимз развития кетоацидоза.
Роль контринсулярных гормонов:
– адреналин, кортизол и СТГ подавляют опосредованную инсулином утилизацию глюкозы мышечной тканью;
– адреналин, глюкагон и кортизол усиливают гликогенолиз и глюконеогенез;
– адреналин и СТГ усиливают липолиз и подавляют остаточную секрецию инсулина.
Клиническая картина ДКА
Самые частые симптомы – полидипсия, полиурия, слабость; выраженность этих симптомов зависит от степени и продолжительности гипергликемии. Больные могут жаловаться на отсутствие аппетита, тошноту, рвоту, связанные с кетонемией боли в животе, которые могут имитировать различные хирургические заболевания. Гипокалиемия и гипокалигистия может вызвать атонию гладкой мускулатуры желудочно-кишечного тракта, и, как следствие – кишечную непроходимость и острое расширение желудка, что приводит к тяжелой рвоте.
Характерна также гипервентиляция за счет увеличения глубины дыхания, при падении рН ниже 7,2 обычно возникает дыхание Куссмауля, сопровождающееся запахом ацетона. Часто возникает тахикардия. При отсутствии выраженной дегидратации гипотония отсутствует. Обычно наблюдается гипотермия. Может отмечаться гипорефлексия, вызванная гипокалиемией. При тяжелом течении диабетического кетоацидоза наблюдаются артериальная гипотония, сопор, кома.
Другая причина кетоацидоза – 2 стадия голодания. Со 2-го – 3-го дня полного голодания в связи с мобилизацией больших количеств жира из депо и образованием кетоновых тел возникает транзиторный кетоз, не требующий лечения, так как в условиях гипогликемии и гипоинсулинемии клетки начинают использовать кетоны в качестве основного энергетического субстрата. Могут проявляться признаки ацидоза – головная боль, головокружение, тошнота, слабость, инсомния. При лечебном голодании в этом случае назначают энтерально щелочные растворы.
Алкогольный кетоацидоз наступает у больных хроническим алкоголизмом после резкого прекращения приема этанола. Обычно он обусловлен рвотой, недостаточным питанием вследствие анорексии, гиповолемией (уменьшением объема внеклеточной жидкости). При острых алкогольных интоксикациях и хроническом алкоголизме также могут активироваться процессы β-окисления жирных кислот. Вследствие увеличения соотношения НАДФ/НАД в крови накапливаются кетоновые тела, преимущественно β-гидроксибутират (по этой же причине возможен незначительный лактоацидоз), который затем превращается в ацетоуксусную кислоту.
Нарушения КОС при метаболическом ацидозе обусловлены перемещением ионов из клетки в интерстициальный сектор и обратно. Более 90% перемещений касаются ионов калия, натрия, хлора, водорода, бикарбоната.
В ответ на повышение концентрации протонов плазмы крови и, соответственно, уменьшение значения рН, избыток протонов связывается бикарбонатным и белковым буферными системами. Ионы водорода перемещаются внутрь клетки, где белковый (в т.ч. гемоглобиновый) буфер является акцептором протонов. Высвобождаемый буфером калий выходит в плазму (феномен биологической травмы клеток), возникает транзиторная гиперкалиемия с сопутствующей прогрессирующей гипокалигистией. Калий интенсивно выводится с мочой, суточная гиперкалийурия может превышать норму в 4-5 раз. За счет этого в течение 4-5 дней при условиях наличия ацидоза содержание калия в плазме нормализуется, затем развивается гипокалиемия.
На каждые 3 иона калия, выходящего из клетки, в нее перемещаются 2 иона натрия и один ион водорода (трансминерализация клетки). Это приводит к усугублению клеточного ацидоза и перестройке ферментных систем. Гипернатриемия вызывает клеточную гипергидратацию.
Механизмы компенсации при метаболическом ацидозе направлены на уменьшение концентрации ионов водорода и осуществляются вне- и внутриклеточными буферными системами, легкими и почками. В плазме снижается содержание бикарбоната за счет истощения бикарбонатного буфера.
Повышение концентрации протонов вызывает стимуляцию дыхательного центра, развивается гипервентиляция и тахипноэ, рСО2 внеклеточной жидкости снижается до тех пор, пока не станет достаточным для выравнивания Н2СО3/НСО3- (при снижении НСО3- на 2 мЭкв/л рСО2 компенсаторно снижается на 1,3 мм рт.ст.). При снижении рН менее 7,2 может развиваться дыхание Куссмауля. Однако дыхательная компенсация не может полностью нормализовать концентрацию протонов, так как именно высокий уровень [Н+] стимулирует дыхательный центр. Кроме того, усиленная работа дыхательной мускулатуры способствует дополнительному образованию углекислого газа. В случае несостоятельности респираторных механизмов (заболевания органов дыхания, нарушения центральной регуляции дыхания и др.) быстрая компенсация невозможна, развивается тяжелый смешанный ацидоз с резким уменьшением рН и почти неизмененным содержанием бикарбоната.
Почечный механизм компенсации метаболического ацидоза включается позднее, спустя 16-18 часов от начала воздействия повреждающего фактора. Механизм направлен на усиление экскреции протонов и играет важную роль только у больных с неизмененной функцией почек. В почках усиливаются процессы ацидо- и аммониогенеза, а также восполнения дефицита бикарбоната плазмы за счет его образования и реабсорбции. Усиленная реабсорбция ионов бикарбоната приводит к активной экскреции хлора (ионы НСО3ˉ Сl- являются реципрокными), развивается гипохлоремия.
В печени активируются процессы дезаминирования аминокислот, вовлекаются в метаболизм лактат и пируват, тем самым уменьшается уровень кислореагирующих продуктов.
Лабораторные показатели метаболического ацидоза
- снижение рН плазмы
- уменьшение значений рСО2, АВ, SВ, ВВ
- сдвиг ВЕ в отрицательную сторону
- резкое снижение рН мочи (ниже 4,0)
- гиперкалиемия (в начальной стадии), сменяющаяся в последующем гипокалиемией
- гипохлоремия
- гипернатриемия
Терапия метаболического ацидоза включает прежде всего устранение причин, приводящих к дыхательной гипоксии (борьба с гиповентиляцией, оксигенотерапия, вспомогательное дыхание, при необходимости – интубация и перевод больного на ИВЛ).
Следующим важным моментом является борьба с расстройствами кровообращения (ликвидация гиповолемии, анемии, улучшение реологических свойств крови, микроциркуляции, лечение сердечной недостаточности).
Внутривенно вводя щелочные растворы: бикарбонат натрия, лактат натрия, лактасол, трисамин, ацесоль. Бикарбонат натрия применяют при тяжелом ацидозе (рН ниже 7,2) для улучшения сократимости миокарда и повышения утилизации лактата. Однако, поскольку бикарбонат натрия активирует фосфофруктокиназу, то при его применении возможно парадоксальное усугубление ацидоза за счет увеличения продукции лактата.
Расчет количества бикарбоната натрия производят по формуле дефицита буферных оснований (ДБО).
ДБО = 0,3 ВЕ масса тела (ммоль)
Вопрос о применении щелочных растворов при умеренном ацидозе до сих пор не решен. Введение бикарбоната натрия может приводить к артериальной гипертензии и отеку легких в связи с повышением тонуса вен и олигурией. Если удается устранить причину ацидоза,.то введение бикарбоната может привести к развитию алкалоза.
До сих пор не решен вопрос о целесообразности применения бикарбоната для устранения ацидоза при ДКА, так как при его использовании возникают нежелательные эффекты:
– При ДКА из-за потери фосфора снижается уровень 2,3-дифосфоглицерата в эритроцитах, и в результате кривая диссоциации оксигемоглобина сдвигается влево (кислород прочнее связывается с гемоглобином). Ацидоз сдвигает кривую диссоциации вправо (эффект Бора), так что снабжение тканей кислородом не нарушается. Бикарбонат вновь сдвигает кривую диссоциации влево, и ткани получают меньше кислорода.
– При правильном лечении (инфузионная терапия плюс инсулин) кетоновые тела превращаются в бикарбонат и введение дополнительного бикарбоната может вызвать алкалоз .
– Имеющиеся в продаже растворы бикарбоната гиперосмолярны и могут увеличивать и без того высокую осмоляльность плазмы.
– Лечение бикарбонатом может вызвать неврологические осложнения - от спутанности сознания до комы . Анионы HCO3-, соединяясь с ионами H+, образуют угольную кислоту . При ее диссоциации образуются CO2 и вода. CO2 легко проникает через гематоэнцефалический барьер и вызывает закисление спинномозговой жидкости (сам бикарбонат в нее практически не проникает ). Поэтому нормализация pH плазмы может сопровождаться парадоксальным ацидозом в ЦНС, который и приводит к неврологическим нарушениям.
Применение бикарбоната оправдано в следущих случаях:
– если ДКА осложняется тяжелым лактацидозом ;
– если имеется тяжелый ацидоз (pH менее 6,9), особенно осложненный шоком, который не поддается инфузионной терапии, направленной на повышение сердечного выброса (0,9% раствор NaCl, плазма, альбумин).
При повышении pH до 7,1-7,15 бикарбонат отменяют.
Трисамин используется при избытке в крови натрия и неизменной функци почек. Эффект трисамина основан на свободном проникновении препарата в клетку и коррекции внутриклеточного ацидоза. Имеет побочные дейсвия: способствует миграции калия из клетки, может вызвать гипогликемию и гипокальциемию.
Расчет производят по формуле:
Кол-во р-ра= масса тела (кг) ВЕ
Реже используется лактат. Его использование будет эффективным, если у больного сохранены окислительные процессы в клетке. Если имеется гипоксия, то в крови будет накапливаться молочная кислота.
Одна из важных проблем лечения метаболического ацидоза – коррекция электролитных нарушений, в частности обмена калия и хлора. По мере устранения ацидоза путем инфузионной терапии и введения инсулина при ДКА калий поступает обратно в клетки и его уровень в сыворотке снижается, что может вызвать гипокалиемию. Гипокалиемия вызывает тяжелые осложнения (например, угрожающие жизни желудочковые аритмии). Если нет задержки мочи, восполнение калия начинают одновременно с инфузионной терапией. Если уровень калия исходно повышен, его назначают только после того, как его концентрация в сыворотке нормализуется. Дозы калия корректируют на основании данных ЭКГ и клинических и лабораторных показателей гипокалиемии
У мужчины 22 лет, в течение 15 лет страдающего инсулин-зависимым сахарным диабетом, в течение трех дней были тошнота и рвота. Он жаловался на выраженную жажду и частое мочеиспускание. У пациента наблюдалось дыхание Куссмауля.
рН 7,15
рО2 94 мм Hg
рСО2 25 мм Hg
SB 6,8 ммоль/л
ВВ 25,0 ммоль/л
ВЕ -23,0
Уровень глюкозы в крови 23,0 ммоль/л.
Клинические и лабораторные данные типичны для диабетического кетоацидоза в стадии декомпенсации. Низкая концентрация бикарбоната, выраженный сдвиг буферных оснований в отрицательную сторону, гипервентиляция с резким снижением рСО2 свидетельствуют о наличии метаболического ацидоза с частичной дыхательной компенсацией.
Экзогенный ацидоз
Экзогенный ацидозвозникает при употреблении некоторых лекарственных препаратов и отравлениях. Его причины: переливание большого количества растворов с рН ниже 7,0; отравления салицилатами, хлоралгидратом, суррогатами алкоголя, этиленгликолем; длительное употребление кислых продуктов. Действие салицилатов состоит в образовании метаболического блока, приводящего к появлению смеси эндогенных органических кислот. Метанол и этиленгликоль превращаются в кислые метаболиты, метанол – в муравьиную кислоту, этиленгликоль – в глиоксиловую и щавелевую кислоты. Отравление метиловым спиртом и салицилатами активирует также продукцию молочной кислоты, способствуя развитию лактацидоза. Некоторые сахара, применяемые для парентерального питания (фруктоза), также могут вызвать лактацидоз. Следует заметить, что большинство растворов, применяемых в трансфузионной практике, также имеют кислую реакцию и могут оказывать влияние на рН плазмы.
| рН | |
| Глюкоза | 3,5–4,0 |
| Полиглюкин | 4,6–5,0 |
| Реополиглюкин | 4,0–6,2 |
| Поливинол | 5,2–5,8 |
| Аминопептид | 5,7–6,7 |
| Казеин | 5,7–6,7 |
| Физиологический раствор | 6,6–7,0 |
| Белковый гидролизат | 6,4–6,5 |
| Гемодез | 5,2–7,0 |
| Аминокровин | 6,4–6,7 |
| Консервированная кровь | 6,5–7,0 |
| Желатиноль | 6,7–7,2 |
Токсичность салицилатов. Салицилаты являются потенциальным источником метаболических кислот. Аспирин (ацетилсалициловая кислота) быстро превращается в организме в салициловую кислоту. Он чаще, чем другие препараты салициловой кислоты, приводит к развитию салицилатной интоксикации. Большие дозы салицилатов вызывают серьезные токсические эффекты. Фатальная доза может быть как небольшой, так и значительной (от 10 до 30 г у взрослых и 3 г у детей).
При салицилатной интоксикации встречаются разнообразные нарушения кислотно-основного состояния. Салицилаты проникают через гемато-энцефалический барьер и напрямую стимулируют дыхательный центр, вызывая гипервентиляцию и респираторный алкалоз. Почки вступают в компенсацию, секретируя повышенные количества НСО3ˉ , К+ и Na+, внося, таким образом, вклад в развитие метаболического ацидоза. Салицилаты также препятствуют метаболизму углеводов, что приводит к повышению продукции метаболических кислот.
Для лечения интоксикации салицилатами используется ощелачивание плазмы. Салициловая кислота, являющаяся слабой кислотой, находится в равновесии с основным (щелочным) салицилатным ионом. Токсичность салициловой кислоты связана с ее способностью проникать через мембраны и входить в клетки мозга. Для салицилатного аниона клеточные мембраны менее проницаема, что и определяет меньшую токсичность. При ощелачивании внеклеточной жидкости, отношение салициловой кислоты к салицилату существенно уменьшается, приводя к перемещению ацетилсалициловой кислоты из клеток во внеклеточную жидкость по градиенту концентрации. Почечная элиминация салицилатов следует подобному паттерну при защелачивании мочи.
Интоксикация метанолом и этиленгликолем. Накопление метанола и этиленгликоля в организме приводит к образованию метаболических кислот и вызывает метаболический ацидоз. И метанол, и этиленгликоль увеличивают анионный интервал.
Метанол (древесный алкоголь) является компонентом шеллака (природного лака), лака, антифризов и других коммерческих продуктов. Метанол может абсорбироваться через кожу, всасываться в желудочно-кишечном тракте либо поступать ингаляционным путем через легкие. Летальной может быть как небольшая, так и значительная доза, не превышающая 30 мл. Кроме способности вызывать метаболический ацидоз, метанол проявляет токсичность в отношении зрительного нерва и центральной нервной системы. Системные органные повреждения возникают через 24 часа, в течение которых метанол превращается в формальдегид и муравьиную кислоту.
Этиленгликоль – растворитель, содержащийся в большом количестве промышленных товаров: от антифризов до очищающих средств (ковровых покрытий, полов). Летальная доза составляет приблизительно 100 мл. Этиленгликоль быстро абсорбируется из кишечника, делая лечение с помощью промывания желудка неэффективным. Ацидоз возникает вследствие превращения этиленгликоля в организме в щавелевую и молочную кислоту. Проявления интоксикации этиленгликолем многообразны: неврологические симптомы (от возбуждения до комы), которые появляются в первые 12 часов; кардио-респираторные нарушения, такие как тахикардия и отек легких; боль в брюшной полости, развитие почечной недостаточности, вызванной закупориванием канальцев кристаллами оксалатов (вследствие повышенной продукции солей щавелевой кислоты).
Фермент алкогольдегидрогененаза превращает метанол и этиленгликоль в токсичные метаболиты. Это тот же фермент, который принимает участие в метаболизме этанола. Поскольку аффинность алкогольдегидрогеназы к этанолу в 10 раз превышает сродство к метанолу и этиленгликолю, внутривенное или пероральное использование этанола как антидота при возникновении отравления метанолом и этиленгликолем вполне оправдано. Также используются инфузионная терапия (для увеличения объема внеклеточной жидкости) и гемодиализ.
Выделительный ацидоз
Выделительный ацидоз развивается при нарушении процессов ацидо- и аммониогенеза в почках или при избыточной потере оснований с каловыми массами.
Причины кишечного выделительного ацидоза:
1. Диарея
2. Уретеросигмоидостомия
Тяжелая диарея или нарушение процессов всасывания в тонкой кишке вызывает развитие ацидоза вследствие потери бикарбоната с каловыми массами (40-60 ммоль/л). Уретеросигмоидостомия, т.е. наложение анастомоза между мочеточниками и сигмовидной кишкой, ведет к метаболическому ацидозу вследствие обмена ионов хлора на ионы бикарбоната в эпителии тонкой кишки и частого развития после операции заболеваний почек (обструктивная уропатия и пиелонефрит).
Почечный выделительный ацидоз развивается при заболеваниях почек, которые сопровождаются нарушениями механизмов секреции в канальцах ионов водорода и реабсорбции натрия и бикарбоната.
Ацидоз при хронической почечной недостаточности связан со снижением способности почек экскретировать аммиак, с гипонатриемией, гипохлоремией, дегидратацией, возникающими при рвоте и диарее. У больных также может увеличиваться экскреция бикарбоната, однако его концентрация обычно стабилизируется на уровне 12-18 ммоль/л. Даже в терминальных стадиях ХПН количество бикарбоната редко снижается менее 10 ммоль/л. Это может объясняться тем, что во-первых, ацидоз стимулирует экскрецию кислоты, во-вторых, карбонат и фосфат костной ткани оказывают буферное действие метаболическим кислотам. Почечный выделительный ацидоз чаще встречается при пиелонефрите вследствие поражения канальцев; при гломерулонефрите он развивается при падении СКФ ниже 30-40 мл/мин.
При любом патологическом процессе, повреждающем почечные канальцы, нарушен ацидогенез и реабсорбция и образование бикарбонатов. Развивается канальцевый ацидоз. В соответствии с причинами выделяют:
– Проксимальный канальцевый ацидоз. Он характеризуется снижением проксимальной канальцевой реабсорбции бикарбоната, приводящей к чрезмерной потере его с мочой. Причины – цистиноз, системная красная волчанка, множественая миелома, отравление солями тяжелых металлов, болезнь Уилсона, нефротический синдром, синдром Фанкони. Для этого вида ацидоза характерны глюкозурия, аминоацидурия, фосфатурия, гипокалиемия.
– Дистальный канальцевый ацидоз. Он возникает вследствие снижения секреции протонов в дистальных отделах канальцев и уменьшения образования бикарбоната. Причины – цистиноз, СКВ, множественная миелома, обструктивная уропатия, синдром Шегрена, длительный прием некоторых медикаментов (анальгетики, диуретики – ингибиторы карбоангидразы, бисептол, противосудорожные препараты, ифосфамид, препарат, применяемый для лечения цитомегаловирусной инфекции – фоскариет). Для этого типа ацидоза характерны глюкозурия, аминоацидурия, фосфатурия, гипокалиемия. Вследствие снижения секреции ионов водорода уменьшения рН мочи не происходит.
– Дистальный канальцевый гиперкалиемический ацидоз. Развивается при блокаде бикарбонат-реабсорбционной функции почек и увеличением рН мочи за счет щелочного резерва плазмы, нарущении экскреции протонов и калия вледствие дефицита минералкортикоидов (болезнь Аддисона, действие гепарин-сульфата) или снижения чувствительности к ним при хронической почечной недостаточности. Сопровождается уменьшением образования NН3, снижением секреции протонов, нарушением образования бикарбоната, гиперкалиемией, сдвигом рН мочи в щелочную сторону.
Почечный выделительный ацидоз может развиваться при применении диуретика ацетазоламида (диакарб), ингибирующего карбоангидразу и уменьшающего проксимальную канальцевую реабсорбцию бикарбоната.
Анионный интервал в дифференциальной диагностике нереспираторного ацидоза
| Снижение анионного интервала (<8 mEq/L) Гипоальбуминемия (снижение неизмеряемых анионов) Множественная миелома (увеличение неизмеряемого катионного парапротеина IgG) Повышение неизмеряемых катионов (гиперкалиемия, гиперкальциемия, гипермагниемия, интоксикация литием) |
| Повышение анионного интервала (>12 mEq/L) Присутствие неизмеряемых метаболических анионов Диабетический кетоацидоз Алкогольный кетоацидоз Молочно-кислый ацидоз Голодание Почечная недостаточность Присутствие лекарственных или химических анионов Интоксикация салицилатами Интоксикация метанолом Интоксикация этиленгликолем |
| Нормальный анионный интервал (8-12 mEq/L) Потеря бикарбонатов Диарея Потеря панкреатического сока Илеостомия (неадаптированная) Накопление хлоридов Почечный канальцевый ацидоз Парентеральное питание (аргинин и лизин) |
Респираторный (дыхательный, газовый) ацидоз
Респираторный ацидоз развивается при накоплении СО2, ведущем к повышению концентрации угольной кислоты. Наблюдается при альвеолярной гиповентиляции. Снижение вентиляции может быть результатом снижения стимуляции дыхания, болезней легких, нарушений со стороны грудной клетки и дыхательных мышц. Реже респираторный ацидоз возникает вследствие повышенной продукции СО2.
Острые нарушения вентиляции. Острый респираторный ацидоз может быть вызван снижением функции дыхательного центра в мозге (например, при передозировке опиатов, барбитуратов), заболеваниями легких, повреждением грудной клетки, слабостью дыхательных мышц или обструкцией воздухопроводящих путей. У большинства пациентов с острым респираторным ацидозом развивается гипоксемия при вдыхании воздуха, имеющего обычный состав. Во многих случаях признаки гипоксемии развиваются до появления признаков респираторного ацидоза, поскольку СО2 диффундирует через альвеолярно-капиллярную мембрану в 20 раз быстрее кислорода.
Хронические нарушения вентиляции. Хронический респираторный ацидоз характерен для пациентов с хронической обструктивной болезнью легких. У этих лиц продолжительный подъем СО2 в крови стимулирует секрецию ионов Н+ и реабсорбцию НСО3ˉ ионов в почечных канальцах. Благодаря эффективности этих компенсаторных механизмов рН возвращается к нормальным значениям до тех пор, пока уровень кислорода поддерживается в пределах, не подавляющих хеморецепторный контроль дыхания.
Острые эпизоды респираторного ацидоза могут развиваться у пациентов с хронической болезнью легких, имеющих постоянно повышенный уровень рСО2. Это состояние иногда называют углекислым наркозом. У этих пациентов дыхательный центр адаптируется к повышенным уровням СО2. У больных ХОБЛ содержание кислорода в крови (вместо рСО2) становится главным стимулом, регулирующим дыхание. При применении кислородотерапии частота и глубина дыхания снижаются, а содержание СО2 в крови возрастает.
Повышенная продукция двуокиси углерода. Двуокись углерода является продуктом метаболических процессов организма, образующих значительные количества кислот, которые должны быть выведены легкими и почками, чтобы предотвратить развитие ацидоза. Продукция СО2 повышается приблизительно на 13% при подъеме температуры выше нормальной на каждый 10С. Еда также индуцирует продукцию СО2. Богатая углеводами диета проводит к образованию более значительному количеству СО2, чем пища, содержащая рациональные количества протеинов и жиров. Хотя избыточная продукция двуокиси углерода может приводить к повышению рСО2, однако у здоровых людей это случается редко, поскольку повышение СО2 обычно сопряжено с соответствующим выведением двуокиси углерода легкими. У пациентов с заболеваниями легких респираторный ацидоз развивается из-за затруднения элиминации избытка СО2 с выдыхаемым воздухом.
Причины респираторного ацидоза:
– Нарушение центральной регуляции дыхания: дисфункция или травма синокаротидной зоны, длительная гипоксия, бульбарная форма полиомиелита или энцефалита, инфаркт, кровоизлияние или травма ствола мозга, демиелинезирующее или дегенеративное поражение ствола мозга, прием некоторых лекарственных препаратов (опиаты, барбитураты и др.).
– Поражения дыхательных мышц или нарушения их иннервации: травма верхнешейного отдела позвоночника, полиомиелит, болезни мотонейронов, нейропатии, миастении, миопатии, лечение миорелаксантами.
– Нарушение подвижности грудной клетки: кифосколиоз, анкилозирующий спондилит, ожирение и т.д.
– Нарушение функции органов дыхания: хронические обструктивные заболевания легких, пневмония, бронхиальная астма, стеноз гортани и трахеи, муковисцидоз, ТЭЛА, эмфизема, пневмоторакс, отек легких, респираторный дистресс-синдром и т.д
– Неправильно подобранный режим ИВЛ.
Эти причины приводят к развитию гиперкапнии и гипоксии, что усугубляет ацидоз за счет присоединения метаболического компонента. Поскольку скорость метаболической продукции углекислого газа высока, недостаточность легочной вентиляции быстро приводит к повышению рСО2. При стойкой гиперкапнии начинает усиливаться экскреция протонов в почках в основном за счет ионов аммония), что приводит к активации механизма реабсорбции бикарбоната, уровень его в плазме растет. Концентрация бикарбоната в плазме увеличивается со скоростью 3 ммоль/л на каждые 10 мм рт.ст. повышения рСО2, тем самым компенсируя снижение рН плазмы. Возрастание реабсорбции бикарбоната сопровождается усилением секреции хлора, что приводит к гипохлоремии.
В норме повышение рСО2 стимулирует легочную вентиляцию, направленную на устранение гиперкапнии. Однако значительное повышение уровня углекислого газа начинает оказывать угнетающее действие на центральную нервную систему. Респираторный ацидоз всегда сопровождается гипоксией. Почечные механизмы компенсации (секреция протонов и реабсорбция бикарбоната) действуют слишком медленно, и рН может восстановиться только через несколько дней.
Клинические проявления респираторного ацидоза: одышка, слабость, головная боль, бессонница, цианоз, тахипноэ. По мере прогрессирования ацидоза беспокойство сменяется сопорозным состоянием. Характерно расширение поверхностных сосудов лица и конъюнктивы. Гиперкапния и гипоксия вызывают гиперкатехоламинемию, повышается тонус артериол, развивается гипертензия. При прогрессировании нарастает тканевая гипоксия, респираторный ацидоз усугубляется метаболическим. Снижается чувствительность адренорецепторов к катехоламинам. Прогрессирует сердечная недостаточность, возникает гипотензия, легочная гипертензия.
Клиническими признаками легкой гиперкапнии (рСО2 46-80 мм рт.ст.) являются легкая гиперемия лица и слизистых, повышение температуры кожных покровов, потливость, подъем АД, олигурия. Указанные признаки нередко расцениваются как положительное явление, в действительности это состояние ложного благополучия.
При умеренной гиперкапнии (рСО2 80-100мм рт.ст.) отмечаются выраженные патофизиологические изменения со стороны органов дыхания, кровообращения, ЦНС. Возможны аритмия, артериальная гипотония, нарушения сознания, тяжелый ацидоз, гиперкалиемия.
При выраженной гиперкапнии (рСО2 200мм рт.ст. и более) наступают глубокий газовый ацидоз, коматозное состояние, брадикардия, аритмия, остановка кровообращения.
Механизмы компенсации при респираторном ацидозе направлены на ликвидацию гиперкапнии и нейтрализацию избытка протонов плазмы. Увеличение рСО2 стимулирует легочную вентиляцию. Избыток ионов водорода плазмы нейтрализуется бикарбонатной и белковой буферными системами. В почках происходит форсированное выведение Н+, рН мочи сдвигается в кислую сторону. В клетках почечных канальцев повышается активность карбоангидразы, вследствие этого усиливается образование и реабсорбция бикарбоната. Усиленная реабсорбция бикарбоната вызывает увеличение секреции почками ионов хлора, развивается гипохлоремия.
Нейтрализация гемоглобиновым буфером протонов вызывает транзиторную гиперкалиемию (см. Метаболический ацидоз), которая также может продолжаться 4 - 5 суток и затем сменяется гипокалиемией.
На фоне гипоксической гипоксии и газового ацидоза в тканях вторично возникает тканевая гипоксия и метаболический ацидоз. Чувствительность гемоглобина к кислороду в условиях гиперкапнии снижается, уменьшается количество оксигемоглобина, присоединяется гемическая гипоксия, усугубляющая тканевую.
Компенсация газового ацидоза возможна лишь при постепенном его возникновении за счет повышенной экскреции ионов водорода клетками канальцевого эпителия (аммониогенез, выделение кислого фосфата, реабсорбция гидрокарбоната), обмена ионов бикарбоната и хлора между эритроцитом и плазмой с образованием в плазме бикарбоната, а в эритроцитах хлорида калия.
Терапия:
При остром газовом ацидозе механизмы компенсации весьма ограничены. В связи с этим интенсивная терапия газового ацидоза состоит в устранении причин гиповентиляции и при соответствующих показаниях – переводе больного на ИВЛ. Необходимо также оптимизировать периферическое кровообращение и произвести коррекцию метаболических процессов в тканях.
Лабораторные показатели:
- снижение рН плазмы
- увеличение значений рСО2, АВ, SВ, ВВ
- снижение рО2
- умеренный сдвиг ВЕ в положительную сторону
- снижение рН мочи
- гиперкалиемия (в начальной стадии), сменяющаяся в последующем гипокалиемией
- гипохлоремия
- гипернатриемия
Пример респираторного ацидоза
Женщина 31 года приняла неизвестное количество трициклических антидепрессантов. Она была обнаружена без сознания и доставлена в приемный покой с медленным поверхностным дыханием и цианозом. Больная без сознания.
Показатели КОС:
рН 7,17
рО2 65 мм Hg
рСО2 60 мм Hg
SB 21,0 ммоль/л
ВВ 39,5 ммоль/л
ВЕ +8,5 ммоль/л
У больной декомпенсированный респираторный ацидоз с выраженной гиперкапнией, возникший вследствие угнетения дыхательного центра. Значительное снижение рН свидетельствует об отсутствии почечной компенсации, которая может возникнуть на 2-е сутки.
Дата добавления: 2018-02-15; просмотров: 1886; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!