Нейрофибромотоз I– го типа (НФ1)
Синдром Марфана
Наследственная болезнь соединительной ткани
Генетическое нарушение: гетерозиготная мутация в гене фибриллина (FBN1). Локализация в хромосоме 15 q 21.
Тип наследования: аутосомно – доминантный (в 30% случаев – de novo мутации).
Частота заболевания: 1:10000 – 1:15000
Возраст манифестации: с рождения
Основные клинические проявления: симптоматика синдрома Марфана многосистемная и моногообразня: от легких форм, трудноотличимых от нормы, до инвалидизирующего течения. Наиболее специфичными для синдрома Марфана являются нарушение скелета, вывих хрусталика, сердечно - сосудистые изменения, эктазия твердой мозговой оболочки. Диагностические критерии синдрома Марфана должны строго учитываться, поскольку ряд других врожденных дисплазий могут быть приняты за него.
1. Мышечно – скелетная система: арахнодактилия, долихостеномелия, высокий рост, длинные конечности, деформация позвоночника (сколиоз, грудной лордоз, гиперкифоз), деформация передней стенки грудной клетки (вдавленная грудь,“куриная грудь“ или оба варианта), ненормальная подвижность суставов( гиперподвижность, врожденные контрактуры или оба варианта), плоская стопа, высокое арковидное небо, недоразвитость вертлужной впадины, мышечная гипотония.
2. Глаза: вывих хрусталика, миопия, отслоение сетчатки, большая роговица, удлиненная ось глазного яблока, уплощение роговицы.
3. Сердечно – сосудистая система:аортальная регургитация, аневризма восходящей части аорты, расслоение аорты, митральная регургитация, застойные сердечные нарушения, пролапс митрального клапана, кальцификация митрального отверстия, аритмия.
4. Наружные покровы:паховые грыжи, атрофичиские стрии.
5. Легочная система: спонтанный пневмоторакс.
6. Нервная система:экстазия твердой мозговой оболочки, включая пояснично – крестцовое менингоцеле, аномалии развития нервной системы.
Диагностика: диагноз синдрома Марфана основывается на семейном анамнезе, наличии у больного типичных диагностических признаков по результатам физикального осмотра, ЭКГ и ЭхоКГ, офтальмологического и рентгенологического обследования, молекулярно-генетического анализа и лабораторных исследований.
При несомненном синдроме Марфана у родственника 1 степени родства этот диагноз можно установить у пробанда, имеющего проявления болезни в двух системах (органах) и более. Если нет болезни у родственников 1 степени диагноз устанавливается при обнаружении у пробанда нарушений скелета и вовлечения в патологический процесс по меньшей мере двух систем с наиболее специфическими проявлениями в одной из них.
ЭКГ при синдроме Марфана позволяет определить нарушение ритма сердца, выраженную гипертрофию миокарда; ЭхоКГ - обнаружить клапанную регургитацию, увеличение размеров левого желудочка, пролапс митрального клапана, разрывы хорд, дилатацию аорты. На рентгенографии грудной клетки можно увидеть расширение корня и дуги аорты, увеличение размеров сердца; на КТ и МРТ сердца и сосудов - выявить дилатацию и аневризмы аорты.
При синдроме Марфана определяется возрастание (в 2 раза и более) почечной экскреции метаболитов соединительной ткани в моче: глюкозоаминогликанов (и их фракций) и оксипролина. Метод прямого автоматического секвенирования ДНК позволяет провести генетическую идентификацию мутаций в гене FBN1.
Дополнительные сведения: во время беременности увеличивается риск расслоения и разрыва аорты.
Синдром Элерса – Данло
Наследственная болезнь соединительной ткани с разными типами наследования
Генетическое нарушение: мутация в генах, отвечающих за структуру коллагена, или в ферментах метаболизма коллагена. В частности, классические формы (типы 1 и 2) наиболее часто вызваны гетерозиготными мутациями в генах COL 5 A 1 и COL 5 A 2, а сосудистый тип (4) связан с гетерозиготными мутациями в гене COL 3 A 1, кодирующий коллаген типа 3.
Тип наследования: аутосомно – доминантный (в редких случаях: аутосомно – рецессивный и X – сцепленный рецессивный).
Частота заболевания: 1:5000
Возраст манифестации: варьирует
Основные клинические проявления: гиперэластичность и хрупкость кожи, гиперподвижность суставов, склонность к кровотечениям, подверженность травмам(вывихи, растяжения). Клинически, биохимически, молекулярно - генетически выделяют 10 типов синдрома, которые должны считаться самостоятельными нозологическими формами Их симптомы перекрываются, поэтому целесообразно познакомиться со всем комплексом признаков.
1. Кожа: сверхрастяжимость, бархатистость, хрупкость, кровоточивость, темно – коричневые веснушки, рубцы, стрии в области поясницы, просвечивающие вены, расхождение послеоперационных швов.
2. C уставы: пассивное разгибание мизинца на 90 и более градусов, приведение большого пальца кисти к предплечию, переразгибание локтевого сустава на 10 градусов и более, переразгибаиние коленного сустава на 10 градусов и более.
3. Глаза: птоз, избыточное развитие периорбитальной клетчатки, отслойка сетчатки, остатки эпиканта, разрыв глазного яблока.
4. Уши: сверхрастяжимость.
5. Зубы: частичная адентия, сверхкомплектация зубов, опалесцирующая эмаль, пародонтоз, множественный кариес.
6. Грудная клетка: сколиоз, кифоз, лордоз, плоская спина, вдавление грудины.
7. Живот: грыжи, спонтанная перфорация кишечника.
8. Конечности: варикозное расширение вен, подкожные подвижные узелки на голенях, плоскостопие.
9. Сердце: пролапс митрального клапана, аритмии, ВСД.
10. Внутренние органы: птоз желудка, матки и почек + стремительные роды.
11. Мозг: аневризмы сосудов мозга, субарахноидальное кровотечение.
Наиболее диагностически важными признаками являются гиперэластичность кожи, подкожные узелки (сферулы, легче прощуповыемы е на передней поверхности голени), переразгибание суставов, повышенная ранимость тканей, симптомы геморралгического диатеза, пролапс митрального клапана.
В современном варианте классификации синдрома Элерса-Данлоса рассматривается 7 основных типов заболевания:
Классический (типы I и II)
Гипермобильный (тип III)
Сосудистый (тип IV)
Кифосколиоз (тип VI)
Артроклазия (тип VII A, B)
Дермоспараксис (тип VIIC)
Недостаток тенасцина-X
I тип синдрома Элерса-Данлоса (классический тяжелого течения) – наиболее частый вариант заболевания (43% случаев) с аутосомно-доминантным типом наследования. Ведущим симптомом является гиперэластичность кожи, растяжимость которой по сравнению с нормой увеличена в 2-2,5 раза. Характерна гипермобильность суставов, носящая генерализованный характер, деформации скелета, повышенная ранимость кожи, склонность к наружным кровотечениям, образованию рубцов, плохому заживлению ран. У части больных выявляется наличие моллюскоподобных псевдоопухолей и варикозного расширения вен нижних конечностей. Беременность у женщин с I типом синдрома Элерса-Данлоса часто осложняется преждевременными родами.
II тип синдрома Элерса-Данлоса (классический мягкого течения) – характеризуется вышеописанными признаками, но выраженными в меньшей степени. Растяжимость кожи превосходит нормальную лишь на 30%; гипермобильность отмечается преимущественно в суставах стоп и кистей; кровоточивость и наклонность к рубцеванию незначительны.
III тип синдрома Элерса-Данлоса – имеет аутосомно-доминантное наследование, доброкачественное течение. Клинические проявления включают генерализованную повышенную подвижность суставов, скелетно-мышечные деформации. Остальные проявления (гиперэластичность и рубцевание кожи, геморрагии) минимальны.
IV тип синдрома Элерса-Данлоса – встречается редко, протекает тяжело; может наследоваться различными путями (доминантно или рецессивно). Гиперэластичность кожи незначительна, отмечается повышенная подвижность только суставов пальцев рук. Ведущим проявлением данного типа заболевания является геморрагический синдром: склонность к образованию экхимозов, спонтанных гематом (в т. ч. во внутренних органах), разрывам полых органов и сосудов (в т. ч. аорты). Сопровождается высокой летальностью.
V тип синдрома Элерса-Данлоса – имеет Х-сцепленное рецессивное наследование. Характеризуется повышенной растяжимостью кожи, умеренно выраженными гипермобильностью суставов, кровоточивостью и ранимостью кожи.
VI тип синдрома Элерса-Данлоса - наследуется по аутосомно-рецессивному типу. Кроме гиперэластичности кожи, наклонности к кровотечениям, повышенной подвижности суставов, имеются мышечная гипотония, тяжелый кифосколиоз, косолапость. Характерной чертой синдрома Элерса-Данлоса VI типа является глазной синдром, проявляющийся близорукостью, кератоконусом, косоглазием, глаукомой, отслойкой сетчатки и т. д.
VII A , B – аутосомно-доминантно - тяжелая гипермобильность сутавов, врожденный вывих бедра.
VII C - аутосомно-доминантно – очень хрупкая кожа и отвисшая кожа.
VIII - аутосомно-доминантно – гиперчувствительность суставов, хрупкость кожи, периодонтоз с потерей зубов.
IX тип (Х-спепленный вариант вялой кожи) в настоящее время исключен из классификации синдрома Элерса-Данлоса.
X тип синдрома Элерса-Данлоса – характеризуется аутосомно-рецессивным наследованием; умеренной гиперэластичностью кожи и гипермобильностью суставов, стриями (полосовидной атрофией кожи), нарушением агрегации тромбоцитов.
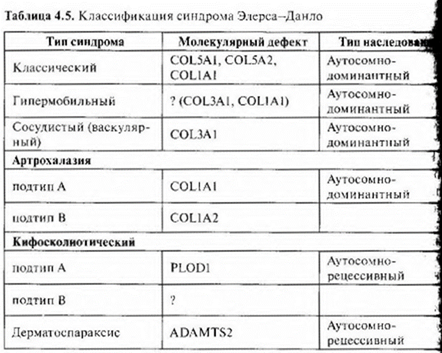
Синдром Элерса—Данло - типичный пример разнолокусной гетерогенности. Все локусы, мутации в которых вызывают синдром, имеют отношение к синтезу белков волокнистых элементов соединительной ткани (главным образом кoллагена). Коллагеновые волока имеют неправильную форму и расположены неупорядочено.
Диагностика: диагностика синдром Элерса-Данлоса проводится медицинским генетиком на основании генеалогических данных, анамнеза, клинического анализа, молекулярно-генетических исследований. Предварительно синдром Элерса-Данлоса может быть заподозрен при наличии больших диагностических критериев (гипермобильности суставов, гиперэластичности кожи, склонности к кровотечениям) и дополнительных малых (хрупкости кожи, патологии сердца, сосудов, глаз и т. д.).
Некоторые формы заболевания требуют проведения биопсии кожи для гистологического, гистохимического, электронно-микроскопического исследования.
Наличие в семье больных синдромом Элерса-Данлоса является показанием к медико-генетическому консультировании и проведению инвазивной пренатальной диагностики.
Больные с различными типами синдром Элерса-Данлоса могут нуждаться в наблюдении и обследовании детским травматологом-ортопедом, детским кардиологом, детским офтальмологом, детским стоматологом, сосудистым хирургом.
ДНК – диагностика.
Дополнительные сведения: наиболее частным типом является III тип, обычно имеющий мягкое течении. Тип IV редок, но опасен осложнениями, связанными с разрывом сосудов и матки у беременных.
Нейрофибромотоз I– го типа (НФ1)
Дата добавления: 2019-07-17; просмотров: 131; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!