Раскрытие необходимости титрования в неводных средах
Титрование в неводных средах- такое титрование, при котором средой служит неводный растворитель с минимальным содержанием воды.
В качестве неводных растворителей применяют обычно обезвоженные (преимущественно органические) жидкости индивидуальные вещества или их смеси, например: ацетон, диметилформамид, диметилсульфоксид, диоксан, кислоты (уксусная, муравьиная), метилэтилкетон, нитрометан, пиридин, спирты (метанол, изопропанол, третичный бутиловый спирт), этилен диамин и др
Используются для определения разнообразных неорганических и органических веществ и для раздельного титрования многокомпонентных смесей солей, кислот и оснований» Благодаря этому возможно титровать:
не только сильные кислоты и основания, но и слабые и очень слабые кислоты и основания;
смеси сильных кислот, смеси сильных оснований; смеси сильных и слабых кислот, смеси сильных и слабых оснований; смеси слабых и очень слабых кислот, смеси слабых и очень слабых оснований;
смеси сильных, слабых и очень слабых кислот; смеси сильных оснований и солей слабых кислот; смеси сильных кислот и солей слабых оснований; смеси свободных и связанных кислот; соли неорганических и органических кислот;
Представление преимуществ и недостатков неводного титрования
Достоинства:
1. Титрование в неводных средах представляет собой очень простой, быстрый и удобный метод количественного анализа многих неорганических, органических и элементорганических соединений.
|
|
|
2. Методы титрования в неводных растворах дают возможность с большой аналитической точностью определять многочисленные вещества, которые при титровании в водной среде не дают резких конечных точек титрования.
3. Одним из важнейших преимуществ методов неводного титрования является то, что они позволяют определять не только растворимые, но и нерастворимые в воде соединения, а также вещества, разлагаемые водой или образующие в водных растворах стойкие нерасслаиваемые эмульсии.
4. Методы титрования в неводных средах могут быть применены для титрования как бесцветных, так и окрашенных растворов.
5. Титрование неводных растворов может проводиться индикаторным, потенциометрическим, кондуктометрическим, амперометрическим и другими физико-химическими методами.
6. При методах неводного титрования во многих случаях не нужно предварительно разделять анализируемые вещества и отделять сопутствующие им примеси или наполнители.
7. Вследствие небольшого, как правило, поверхностного натяжения органических растворителей размеры капель неводных жидкостей меньше размеров капель водных растворов, благодаря чему повышается точность титрования по сравнению с точностью титрования водных растворов.
|
|
|
8. Использование неводных растворителей в аналитической практике дает возможность расширить области их применения в других методах анализа (осаждения, комплексообразования, окисления — восстановления,хроматографии, электрометрических методах и т. д.) и увеличить ассортимент веществ для приготовления титрованных растворов, пригодных для титрования как мономерных, так и полимерных соединений.
Недостатки: долог и надо учитывать, необходимость правильного определения растворителя, чтобы не вносить погрешность.
Расчет кривой титрования действительно нету.
24. Сущность комплексонометрического титрования. Требования к реакциям в комплексонометрии. Комплексоны. Строение ЭДТА. Протолитические и комплексообразующие свойства. Строение комплексонатов. Возможность совместного определения: 1) железа (III) и алюминия; 2) циркония (IV) и цинка.
Комплексонометрия основана на применение реакций образования прочных комплексных соединений с аминополикарбоновыми кислотами.
Комплексонометрическое титрование – метод количественного анализа, основанный на аналитическом использовании реакций комплексообразования с полидентатными хелатообразующими органическими аналитическими реагентами – комплексонами.
|
|
|
указание обоснованных требований, предъявляемым к реакциям
Реакции комплексонообразования для использования в титрометрии должны отвечать общим требованиям: стехиометричность, быстрота, количественность, должна существовать возможность фиксации конечной точки титрования.
Большое значение имеет образование одного продукта реакции с точно определенным хим составом => очевидно преимущество тех реагентов, которые взаимодействуют в 1 стадию, максимум в 2.
представление структурных формул комплексонов
Комплексоныи их формулы:
Нитрилотриуксусная кислота

Этилендиаминтетрауксусная кислота (ЭДТУ)

Двунатриевая соль этилендиаминтетрауксусной кислоты (ЭДТА)

Натриевая соль транс-1,2-диаминциклогексантетрауксусной кислоты

рассмотрение строения молекулы ЭДТА и форм его существования в растворе
ЭДТА - двунатриевая соль этилендиаминтетрауксусной кислоты (комплексон III). Молекула ЭДТА содержит 4 карбоксильные группы и 2 третичных азота из этого следует что ЭДТА одновременно является и кислотой способной давать соли с различными металлами, а так же является комплексообразователем способным давать прочные, растворимые внутрикомплексные соединения. ЭДТА может обр. 4 ковалентные связи с карбоксильными группами и 2 координационные связи с азотом.
|
|
|
ЭДТА в р-ре имеет биполярную бетаиновую структуру. Одновременно содержащую группу четвертичного аммонийного основания и карбоксилат анион. 
представление свойств ЭДТА
Протолитические свойства: две аминодиацетатные группировки придают ЭДТА св-ва слабого электролита при этом состояние молекул ЭДТА в р-ре, а так же её способность обр. комплексы с ионами металлов сильно зависят от pH. В сильнокислых средах (pH ≤0) возможно протонированние атомов азота и обр. катиона Na2HY+ или Na2H2Y2+.
H4Y↔H++H3Y-; H3Y-↔H++H2Y2-; H2Y2-↔H++HY3-; HY3-↔H++Y4-;
В зависимости от кислотности р-ра ЭДТА может быть более или менее протонированна: При pH<1 H4Y; при pH=4,5 H2Y2-; при pH=8,9 HY3-; при pH=>12 Y4-.
Комплексообразующие свойства: ЭДТА имеет 6 функциональных групп – гексадентатный лиганд. ЭДТА обр. циклические внутрикомплексные соединения (хелаты). При обр. хелатов Me координируется за счёт не поделённой пары N, а так же за счёт карбоксильных групп. Молекула ЭДТА способна наилучшим образом удовлетворить координационные и геометрические требования ионов Me. При этом координационная сфера иона Ме полностью насыщается. Не зависимо от Ме ЭДТА обр. комплексы состава 1:1. Образование нескольких хелатных циклов объясняет высокую термодинамическую устойчивость комплексов ЭДТА. Количественно хелатный эффект выражается: ХЭ=lgβMY-lgβMl
С двух-, трех- и четырехзаряженными ионами металлов анион Y4- образует тетраэдрические (Ca2+, Mg2+, Ba2+) и октаэдрические комплексы (комплексонаты) состава MYn-4, где n-заряд иона металла.
Возможность совместного определения: железа (III) и алюминия;
Аликвотную часть анализируемого раствора помещают в коническую колбу для титрования и определяют в ней содержание ионов железа (III) путем прямого титрования стандартным: раствором комплексона III в присутствии сульфосалициловой кислоты при рН=2. В этих условиях Al3+; Ca2+; Mg2+ не мешают определению Fe3+.
Оттитровав железо (III), приступают к определению ионов алюминия в той же пробе анализируемого раствора. Ионы алюминия определяют обратным титрованием предварительно введенного избытка раствора комплексона III стандартным раствором соли железа (III) при pH=4,8-5.При этом значении pH раствора ранее образовавшийся комплексонат железа (III) не разрушается и не мешает определению ионов алюминия.
Формула:  а — навеска, г; W — содержание воды в силикате, %.
а — навеска, г; W — содержание воды в силикате, %.
Содержание окиси алюминия рассчитывают по формуле:

где V1ЭДТА — объем стандартного раствора комплексона III, добавленного после титрования Fe3+, мл; V11ЭДТА — объем стандартного раствора соли железа (III), пошедшего на обратное титрование избытка комплексона III, мл; ТЭДТА/ Al2O3 — титр комплексона III, выраженный по определяемому веществу, г/мл Al2O3;
Возможность совместного определения: циркония (IV) и цинка.(по подобию)
25. Комплексонометрическое титрование кальция и магния. Приёмы повышения избирательности комплексонометрического титрования. Постройте кривую титрования кальция в присутствии магния. Факторы, влияющие на вид кривой титрования и величину скачка.
Количественное определение массы кальция в анализируемом растворе проводят методом прямого комплексонометрического титрования в щелочной среде (рН12) в присутствии индикатора мурексидаNH4H4In.
H4In-+2OH-↔2H2O+H2In3-
Ca2++H2In3-↔CaH2In-
CaH2In-+Y4-↔CaY2-+H2In3-
Полученную контрольную задачу в мерной колбе на 25 мл разбавляют дистиллированной водой, доводят объем раствора до метки и перемешивают. В колбу для титрования отбирают мерной пипеткой 1 мл полученного раствора, добавляют 1 мл раствора натрия гидроксида до рН=12 (контроль по универсальной индикаторной бумаге), 2 мл дистиллированной воды, несколько кристаллов индикаторной смеси мурексида до появления розовой окраски раствора и титруют раствором ЭДТА до перехода розовой окраски раствора в фиолетовую (или синюю).
Раздельное определение Са2+ и Mg2+ в воде основано на хорошей растворимостью гидроксида кальция и малой – гидроксида магния, а также возможности определения ионов Са2+ в сильнощелочных средах комплексонометрически с индикатором мурексид. Анализируемую воду переносят в коническую колбу для титрования, к раствору прибавляют 5 мл аммиачного буферного раствора с рН 8 – 10, индикатор эриохромовый черный Т на кончике шпателя и титруют раствором ЭДТА до перехода окраски из малиновой в синюю. В данных условиях с ЭДТА взаимодействуют как ионы кальция, так и ионы магия. Поэтому полученный V1 (ЭДТА)затрачивается на титрование суммы ионов Ca и Mg. Другую аликвоту анализируемой воды переносят в чистую коническую колбу для титрования, к раствору прибавляют 5 мл 2 моль/л раствора гидроксида натрия, индикатор мурексид на кончике шпателя и титруют раствором ЭДТА до перехода окраски из розовой в лиловую. В данных условиях магний переходит в осадок Mg(OH)2, а с ЭДТА взаимодействуют только ионы кальция, поэтому полученный V2 (ЭДТА) затрачивается на титрование ионов Са.
Количество эквивалентов кальция или магния в анализируемой почве (X), ммоль в 100 г, вычисляют по формуле:
X= 
где V - объем раствора трилона Б, израсходованный на титрование кальция или магния, см3;
V1 - объем раствора трилона Б, израсходованный на титрование кальция или магния в холостой пробе, см3;
с - концентрация раствора трилона C(1/2Na2ЭДТА)=0,1, ммоль/см3;
500 - коэффициент пересчета на 100 г почвы;
V2 - объем пробы анализируемой вытяжки, см3.
Массовую долю кальция в почве (Х1) в процентах вычисляют по формуле:
Х1=С•0,020,
где С - количество эквивалентов кальция в анализируемой почве, ммоль в 100 г;
0,020 - коэффициент пересчета в проценты. Массовую долю магния в анализируемой почве (Х2) в процентах вычисляют по формуле:
Х2=С•0,0122,
где С - количество эквивалентов магния в анализируемой почве, ммоль в 100 г;
0,0122 - коэффициент пересчета в проценты.
Приёмы повышения избирательности комплексонометрического титрования:
ЭДТА яв-ся групповым реагентом, не специфичный.
1)Изменение рН среды. При изменении рН среды изменяется  (степень протекания основного процесса комплексообразования для лиганда), при этом изменяется условная константа устойчивости. Для того, чтобы можно было селективно титровать соответствующие металлы в присутствии других необходимо, чтобы эффективные константы устойчивости различаются по крайне мерев 104-105 раз.
(степень протекания основного процесса комплексообразования для лиганда), при этом изменяется условная константа устойчивости. Для того, чтобы можно было селективно титровать соответствующие металлы в присутствии других необходимо, чтобы эффективные константы устойчивости различаются по крайне мерев 104-105 раз.
Катионы щелочных металлов малоустойчивые комплексы и только в сильно щелочных растворах (рН=11-14)
Катионы щелочноземельных металлов образуют достаточно устойчивые комплексы в интервале рН=4-12.
Для катионов переходных металлов III группы область рН образования устойчивых комплексонатов-3,5-4,5
Тяжёлые многозарядные ионы образуют устойчивые комплексы в кислых средах.
2)Реакциикомплексообразования с маскирующими лигандами. Можно связать мешающие катионы в комплексы при данном рН.
3)Степени окисления металлов.
Внекоторых случаях можно проводить селективное титрование, которое невозможно осуществить с использованием ЭДТА, применяя другой комплексон, например можно титровать Ca в присутствии Mg раствором ГЭДТА, но не ЭДТА. Значительно более избирательным, по сравнению с аминополикарбоновыми кислотами, титрантами являются полиамины, которые практически связывают в комплексы атомы только переходных металлов.
Факторы, влияющие на вид кривой титрования:
Конц-ция определяемого компонента и титранта. Чем меньше начальная конц-ция, тем больше значение координаты в левой ветви кривой титрования, тем меньше скачок. Чем больше конц-ция, тем больше погрешность титрования.
Устойчивость. Чем больше константа устойчивости, тем больше скачок.
Побочные продукты и рН раствора
Комплексометрическое определение ионов железа и цинка. Постройте кривую титрования ионов железа (в ацетатном буферном р-ре), цинка (в аммиачном буферном р-ре). Металлохромные индикаторы. Требования к индикаторам. Равновесие индикатора в р-ре. Интервал перехода окраски индикатора.
Определение граммового содержания железа. Ход анализа: исследуемый раствор, содержащий ионы железа, получают в мерную колбу на 100,0 мл, разбавляют до метки водой и перемешивают. В коническую колбу берут аликвоту разбавленного раствора, прибавляют ≈ 80 – 100 мл дистиллированной воды, 4 – 5 капель раствора сульфосалициловой кислоты и титруют раствором ЭДТА до перехода окраски из вишневой в лимонно- желтую.
Fe3++H2Y2-↔FeY-+2H+
Расчет граммового содержания железа. Титрование повторяют несколько раз. Из трех сходящихся результатов (отличающихся не более, чем на 0,1 мл) вычисляют среднее значение объема титранта.
mFe,г=C(1/2 ЭДТА)  KЭДТАМэкв(Fe)P10-3
KЭДТАМэкв(Fe)P10-3
Количественное определение массы цинка в анализируемом растворе проводят методом прямого комплексонометрического титрования в присутствии аммиачного буфера и индикатора эриохрома черного Т.
При титровании протекают реакции:
Zn2++HY3- ↔ZnY2-+H+
NH3+ H+↔ NH4+
Полученную контрольную задачу в мерной колбе на 25 мл разбавляют дистиллированной водой, доводят до метки и перемешивают. В колбу для титрования отбирают мерной пипеткой 2 мл приготовленного раствора соли цинка, добавляют при перемешивании 1 мл аммиачного буферного раствора и вносят на кончике шпателя 4-5 мг индикаторной смеси эриохрома черного Т до появления красно-фиолетовой окраски раствора. Раствор медленно титруют стандартным раствором ЭДТА до появления сине-голубой окраски.
Металлохромный индикатор-это орг. соединение, образующее с ионом металла комплексное соединение невысокой прочности и отличающиеся по окраске от цвета свободного индикатора.
Эрихромовый чёрный Т, натриевая соль 3-окси-4-[(1-оски-2-нафтил)азо]-7-нитронафталин- 1-сульфокислоты

Ксиленовый оранжевый, натриевая соль 3,3-бис-[N,N-ди-(карбоксиметил)-аминометил]-о-крезолсульфофталеина.

Мурексид(аммонийная соль пурпуровой кислоты) Дифениламин


Требования к индикаторам:
Индикатор должен обладать высоким светопоглащением, чтобы окраска даже его небольшого количества была заметна для человеческого глаза. Большие концентрации индикатора могут привести к заметному расходу на него титранта.
Переход окраски должен быть контрастным.
Область перехода окраски должна быть как можно уже.
Изменение цвета индикатора должно происходить резко. Количество протолита, необходимое для изменения цвета индикатора должно быть настолько мало, чтобы не искажать результаты титрования.
Изменение цвета индикатора должно быть обратимым.
Реакция равновесия индикатора в растворе:
Реакция равновесия индикатора в растворе:
HInd+H2O↔Ind-+H3O+
Kα= 
[H3O+]= 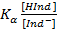
pH=pKα-lg 
 ≥10/1 ≤10/1
≥10/1 ≤10/1
pKα-lg10/1pKα+lg10/1
∆рН=pKα±1 этот интервал называется интервалом перехода окраски индикатора.
27.Комплексометрическое определение меди, алюминия, бария, сульфат-ионов. Способы комплексометрического титрования. Металлохромные индикаторы. Требования к индикаторам. Равновесие индикатора в растворе. Интервал перехода окраски индикатора. Формула для расчёта массового содержания определяемых веществ. Погрешности комплексометрического титрования.
Определение граммового содержания меди. Ход анализа: анализируемый раствор помещают в мерную колбу на 100 мл, доводят водой до метки, тщательно перемешивают. В коническую колбу для титрования берут аликвоту, добавляют индикатор мурексид на кончике шпателя и титруют раствором ЭДТА сначала до грязно-розового цвета, затем добавляют несколько капель 10 %-ного раствора аммиака до появления изумрудной или желтой окраски раствора и дотитровывают раствором ЭДТА до перехода окраски в фиолетовую.
Расчет граммового содержания меди. Титрование повторяют несколько раз. Из трех сходящихся результатов (отличающихся не более, чем на 0,1 мл) вычисляют среднее значение объема титранта.
mCu,г=C(1/2 ЭДТА)· ·KЭДТА·Мэкв(Cu) ·P·10-3
Определение граммового содержания алюминия. Ход анализа:исследуемый раствор помещают в мерную колбу на 100,0 мл, доводят до метки водой и тщательно перемешивают. Аликвоту 20,0 мл переносят в коническую колбу, пипеткой добавляют 25,0 мл 0,0250 моль/л раствора ЭДТА, перемешивают. Затем в колбу для титрования приливают 5 мл аммиачного буферного раствора рН 8 – 10, 70 – 100 мл дистиллированной воды и индикатор эриохромовый черный Т на кончике шпателя. Избыток ЭДТА медленно оттитровывают стандартным 0,0250 моль/л раствором ZnSO4 или MgSO4 до перехода окраски из синей через фиолетовую в малиновую.
Расчет граммового содержания алюминия. Титрование повторяют несколько раз. Из трех сходящихся результатов (отличающихся не более, чем на 0,1 мл) вычисляют среднее значение объема титранта
mAl,г=C(1/2 ЭДТА)·  ·KЭДТА-
·KЭДТА- 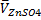 ·KZnSO4) ·Мэкв(Al) ·P·10-3
·KZnSO4) ·Мэкв(Al) ·P·10-3
Комбинируя осаждение с обратным титрованием, можно комплексонометрично определять не только катионы металлов, а и анионы, например, сульфат-,Для этого к исследуемому раствору, который содержит определяемый ион, прибавляют точное количество раствора катиона металла (с известной концентрацией), который образует с анионом осадок. Осадок отфильтровывают. В фильтате определяют избыток катионов стандартным раствором трилона Б.
SO42- + Ba2+ ↔ BaSO4↓
определ. ион избыток
Ba2+ + Н2Y2- ↔ Ba2- + 2Н+
остаток
Ионы Ва2+определят в сильно щелочном р-ре с помочью флуоресцентного индикатора бис [N,K-ди-(карбоксиметил)аминметил]-флуоресцеина, называемого флуорексоном.
Способы комплексометрического титрования:
Прямое титрование-титрование р-ра определяемого вещества непосредственно подходящим –ром титранта.
Обратное титрование заключается в добавлении к определяемому веществу избытка точно известного количества (и точно известной концентрации) стандартного р-ратитранта, после этого оставшийся избыток этого стандартного р-раоттитровывают др. титрантом.
Косвенное титрование. Определяемое вещество непосредственно не реагирует с титрантом, но определяется косвенно в рез-те использования стехиометрическипротекаемой промежуточной реакции его с др. веществом, реагирующим с титрантом.
Титрование по методу вытеснения. Титрование заместителя заключается в титровании не определяемого вещества, а эквивалентного ему заместителя, получающегося в рез-те предварительно проведённой реакции между определяемым веществом и каким-либо вспомогательным реагентом.
Металлохромный индикатор-это орг. соединение, образующее с ионом металла комплексное соединение невысокой прочности и отличающиеся по окраске от цвета свободного индикатора.
Эрихромовый чёрный Т, натриевая соль 3-окси-4-[(1-оски-2-нафтил)азо]-7-нитронафталин- 1-сульфокислоты
Ксиленовый оранжевый, натриевая соль 3,3-бис-[N,N-ди-(карбоксиметил)-аминометил]-о-крезолсульфофталеина.
Мурексид(аммонийная соль пурпуровой кислоты) Дифениламин

Требованияк металлохромным индикаторам:
Комплекс иона металла с индикатором должен быть достаточно устойчив, βMInd>104
Комплексонат иона металла должен быть намного устойчивее комплекса иона металла с индикатором, т.е. βМY/βMind>104
Комплекс иона металла с индикатором должен быть кинетически лабильным и быстро разрушаться при действии ЭДТА
Концентрация индикатора в р-ре должна быть достаточно малой, т.е. С0Ind/C0M<0,01
Изменение окраски индикатора должно быть чётким, контрастным и быстрым.
Реакция равновесия индикатора в растворе:
HInd+H2O↔Ind-+H3O+
Kα=
[H3O+]=
pH=pKα-lg
≥10/1 ≤10/1
pKα-lg10/1pKα+lg10/1
∆рН=pKα±1 этот интервал называется интервалом перехода окраски индикатора.
Погрешности комплексометрического титрования:
ПТ= 
 nY+nMYn0=nM+nMY
nY+nMYn0=nM+nMY
ПТ=  =
=  •100%
•100%
ПТ=  •100%
•100%
ПТ= 
nMY≈n0Y, [MY](VM+VY)≈CMVM
Если рТ>>рМТ  =
= 
Если рТ>>рМТЭ, то величиной  можно пренебречь ПТ=
можно пренебречь ПТ= 
Если рТ<<рМТЭ, то ПТ= 
Сущность окислительно-восстановительного титрования. Условия проведения окислительно-восстановительных реакции. Стандартные и реальные потенциалы. Уравнение Нернста. Факторы, влияющие на величину реального потенциала.
Окислительно-восстановительное титрование(редоксиметрия)-это такие методы анализа, которые используют в качестве титранта р-ры окис-ля и вос-ля.
Используют нагревание для увеличения скорости реакции, в некоторых случаях используют избыток реагента.
Редоксметрический процесс протекает тем полнее, чем больше разность потенциалов двух сопряжённых ОВ пар.
Потенциал возникает в результате перехода  в ходе ОВР, является мерой ОВ способности вещества.
в ходе ОВР, является мерой ОВ способности вещества.
Возможность протекания реакции и глубина определяется разностью потенциалов ЕOx1/Red1> ЕOx2/Red2
Для количественной оценки окислительных или восстановительных св-в системы в р-р погружают электрод из химически инертного токопроводного материала, на границе раздела двух фаз электрод/р-р возникает потенциал, который является функцией активности в р-ре. Значение потенциала тем больше, чем больше окислительная способность. Абсолютно значение потенциала нельзя измерить. Выбирают одну Окислельно-восстановительную систему стандартную и относительно неё измеряют относительный потенциал.
2Н+/Н2 Е=0 при 
Потенциал любой ОВ системы, измеренный в стандартных условиях относительно водородного электрода, называется стандартным потенциалом (Е0) является функцией нескольких величин (ионная сила, константа устойчивости, константа растворимости, константа диссоциации).
Е0 принято считать положительным, если система выступает в качестве окислителя, а на водородном электроде реакция вос-ия. Е0принято считать отрицательным. Если система выстпает в роли вос-ля, на электроде реакция окис-ия.
Для характеристики в реальных условиях используют реальный потенциал. Реальный потенциал отличается от стандартного тем, что зависит от условий, является более ценным. Рассчитывается с помощью уравнения Нернста.
Ox1+ 
E=E0+ 
R=8,314 Дж/моль·К
F=9,6585·104 Кл/моль
E=E0+ 
F=298 K (стандартные условия)
Для разбавленных р-ров и реальных систем в уравнении Нернста С=активности; активность газа=парциальному давлению, Активность твёрдых веществ=1
Факторы, влияющие на величину реального потенциала:
Концентрация или активности окисленных и восстановленных форм. Активность окисленной формы выше, потенциал выше, активность восстановленной формы выше, потенциал ниже.
Т.к.зар. окислительной и восстановительной формы, то ионная сила по-разному влияет.
рН р-ра:
MnO4-+8H++  ↔Mn2++4H2O
↔Mn2++4H2O
Cr2O72-+14 H++  ↔2Cr3++7H2O
↔2Cr3++7H2O
Ox +yH++ 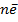 ↔Red
↔Red
E=Е0+  ln
ln 
E=Е0+  lg
lg  pH
pH
Чем ниже рН, тем больше Е окис-й системы, чем выше рН, тем ниже Е.
образование осадка
2Сu2++4I-↔2CuI ↓ +I2
Влияют на потенциал тем сильнее, чем меньше Ks
 =0,54 B
=0,54 B  =0,86 B
=0,86 B
∆ Е0 мала, скорость мала, следовательно, избыток I-
Комплексообразование
Cu-2 =Cu2+
Медная пластинка погружена в 0,01 М р-р соли меди, добавили NH3 и появляется комплекс
Сu2++4 NH3=[Cu(NH3)4]2+
β=[Cu(NH3)42+]/[Cu2+][NH3]4=4,86·1012
 [Cu(NH3)4]2+
[Cu(NH3)4]2+
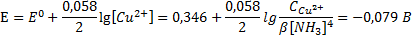
Комплексообразование-это самое мощное средство для изменения Е и, следовательно, направления реакции.
Чем выше температура, тем выше температура, тем выше Е.
Метод определения пероксида водорода в пергидроле, щавелевой кислоты в техническом препарате. Механизм и кинетика данных окислительно -восстновительных реакций. Титранты, индикаторы, стандартизация рабочих р-ров, первичные и вторичные стандарты. Погрешность определения.
Перманганометрический метод определения пероксида водорода состоит в окислении его до свободного кислорода, т.е. здесь используются св-ва пероксида водорода как вос-ля:
5Н2О2+2KMnO4+3H2SO4 →K2SO4+2MnSO4+8H2O+5O2
5О22-+2MnO4-+6H+→2 Mn2++5O2+8H2O
Исследуемый р-рпомещают в мерную колбу, разбавляют дистиллированной водой до метки и перемешивают. Аликвотную часть р-ра переносят в колбу для титрования, разбавляют до 100-120 мл водой, добавляют 20 мл H2SO4(1:4) и титруют р-ром KMnO4до появления слабо- розовой окраски, устойчивой в течение 30 с.
Определение содержания щавелевой кислоты в техническом препарате. Навеску препарата технической щавелевой кислоты растворяют в воде в мерной колбе вместимостью 100 мл. Навеска образца должна быть такой, чтобы после её растворения получился примерно 0.1 моль-экв/л р-р. После растворения и тщательного перемешивания берут аликвоту р-ра пипеткой в коническую колбу для титрования. Добавляют 20 мл воды, 20 мл H2SO4(1:4) и титруют KMnO4до появления розовой окраски, неисчезающей в течение 30-40 с.
2KMnO4+5H2C2O4+3H2SO4→10CO2+2MnSO4+K2SO4+8H2O
2MnO4-+5C2O4 2-+16H+→2 Mn2++10CO2+8H2O
Стандартизация рабочего р-ра:В качестве рабочего р-ра применяется р-р перманганата калия. KMnO4легко получить химически чистым, однако приготовленный р-р в течение 7-10 дней изменит свой титр, поэтому рабочий р-р готовят за ранее. При приготовлении рабочего р-ра необходимо учитывать, для каких определений он будет применяться, т.к. фактор эквивалентности KMnO4в зависимости от условий принимает различные значения.
Для установки коэффициента поправки р-ра перманганата калия используют следующие первичные стандарты: H2C2O4·2H2O, Na2C2O4, (NH4)2C2O4·H2O, K4[Fe(CN)6] ·3H2O, (NH4)2Fe(SO4)2·6H2O, As2O3, металлическое железо и некоторые другие вещества.
Чаще других для стандартизации р-раKMnO4щавелевую кислоту или оксалаты.
В основе определения К поправки KMnO4лежит реакция:
2KMnO4+5Na2C2O4+8H2SO4→10CO2+2MnSO4+K2SO4+8H2O+5Na2SO4
2MnO4-+5C2O4 2-+16H+→2 Mn2++10CO2+8H2O
Навеску оксалата натрия рассчитывают так, чтобы объём р-раKМnO4был равен 15-25 мл. Расчёт ведут по формуле:
m(Na2C2O4)=C(1/5 KMnO4) ·VKMnO4·Mэкв(Na2C2O4) ·10-3
На аналитических весах берут навескуNa2C2O4, помещают ее в коническую колбу для титрования. Навеску растворяют в 120-150 мл воды, добавляют 20 мл H2SO4(1:4), нагревают содержимое колбы до 80 0С и титруют р-ром KMnO4до появления розовой окраски, неисчезающей в течение 30-40 с.

Возможные источники погрешности, формулы для их расчёта:
Использование индикаторов приводит к появлению погрешностей, это связано с несовпадением Е в ТЭ и Е перехода окраски.
Если Е0 IndOх/IndRed< EТЭ
Е0’IndOх/IndRed=Е0’Oх1/Red1+  lg
lg 
ПТ=  =
= 
a= 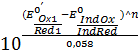
ЕслиЕ0 IndOх/IndRed>EТЭ
Е0’ IndOх/IndRed= Е0’ Oх1/Red1+  lg(f-1)
lg(f-1)
ПТ= 
Перманганатометрическое определение железа (II).КТ сульфата железа (II) в кислом р-ре, содержащем смесь серной и фосфорной кислот. Факторы, влияющие на ход КТ. Условия перманганатометрического определения железа по методу Мора и по методу Циммермана-Рейнгарда. Компоненты смеси Рейенгарда и их роль в процессе титрования железа (II).
Перманганатометрическое титрование применяют преимущественно для определения веществ, играющих по отношению к перманганат-иону роль вос-ля, например металлическое железо. Препарат, содержащий железо, обрабатывают разбавленной серной кислотой при нагревании на водяной бане. Металлическое железо растворяется и переходит в железо(II):Fe + Н2SО4 = FeSO4 + Н2. После растворения железа раствор быстро охлаждают и разбавляют водой. Отбирают аликвоту полученного раствора и определяют железо(II) перманганатометрически прямым титрованием в сернокислой среде на основе реакции:
5Fe2+ + МnО4 ‾ + 8H+ = 5Fe3+ + Mn2+ + 4H2O
Титрование ведут при комнатной температуре до появления устойчивой (в течение ~1—2 минут) розовой окраски раствора.
Поскольку один катион Fe2+ отдает один электрон, то молярная масса эквивалента железа(II) в этой реакции равна его молярной массе.
Аналогично прямым перманганатометрическим титрованием определяют железо(II) в растворах солей железа(II).
При перманганатометрических определениях индикаторы не добавляют, т.к. индикатором является сам перманганат.
Факторы, влияющие на ход КТ:
1)Количество  . Чем больше принимает участие в ОВР, тем более пологая кривая будет наблюдаться.
. Чем больше принимает участие в ОВР, тем более пологая кривая будет наблюдаться.
2)Значение (разница) Е0. Чем больше ЭДС, тем больше скачок.
3)Температура. Повышение температуры увеличивает потенциал и окис-ля и вос-ля, поэтом КТ будет сдвинута по ОУ
4)Разбавление р-ра. Оно влияет, если стехиометрические коэффициенты не равны.
Определение содержания железа в смеси Лунге по методу Мора. В основе определения лежит реакция: 10FeSO4+2KMnO4+8H2SO4 →5Fe2(SO4)3+2MnSO4+K2SO4+ 4H2O. В реакционной смеси Лунге должны содержаться H2SO4 для создания кислой реакции среды, H3PO4 для связывания образующегося в рез-те реакции иона Fe3+ в бесцветный комплекс.
Для определения содержания железа навеску продажного препарата соли Мора помещают в мерную колбу на 100 мл и растворяют в 20 мл конц. H2SO4 и 10 мл конц. H3PO4. В дистиллированной воде соль Мора гидролизуется. Полученный р-р взбалтывают, дают охладиться, добавляют воду до метки и тщательно перемешивают. Аликвотную часть полученного раствора переносят в коническую колбу для титрования, прибавляют 80-100 мл дистиллированной воды, и титруют рабочим р-ром KMnO4 до появления слабо-розовой окраски, устойчивой в течение 30 с.
Определение содержания железа по методу Циммермана-Рейнгарда. Смесь Циммермана-Рейнгарда (67 г MnSO4 ·4H2O, 133 конц. H2SO4 и 160 мл 85% р-раH3PO4, смесь разбавляется водой до 1 л). Навеску анализируемой пробы 0.1500-0.2000 г переносят в коническую колбу и растворяют в 20 мл 10%-ного р-раHCl при нагревании. После чего по каплям прибавляют р-р SnCl2 до обесцвечивания окраски иона Fe3+ : 2FeCl3 +2SnСl2+2HCl→H2SnCl6+2FeCl2
Избыток SnCl2не должен привышать 1-2 капли, его перед титрованием окисляют добавлением 5 мл 5%-ного р-раHgCl2: SnCl2+ 2HgCl2+2HCl→ H2SnCl6+Hg2Cl2.
Чтобы можно было установить момент, когда прибавлено достаточное количество хлорида олова, р-р хлорида железа перед восстановлением нагревают почти до кипячения, интенсивность жёлтой окраски усиливается.Признаком полноты восстановления железа является исчезновение жёлтой окраски р-ра, обесцвечивание горячего р-ра от привления избытка 1-2 капель р-ра хлорида олова хорошо заметно.
Полученный р-р охлаждают, переносят в мерную колбу на 100 мл, добавляют 15 мл смеси Циммермана-Рейнгарда, разводят водой до метки. Аликвотную часть р-ра помещают в коническую колбу для титрования и титруют до слабо-розовой окраски р-ром KMnO4.CмесьЦиммермана-Рейнгардасостоит из MnSO4, H2SO4 и H3PO4. Сульфат марганца обеспечивает понижение потенциала системы Mn(III)/Mn(II), что предотвращает окисление хлорид-иона и создаёт возможность тировать в присутствии его большого избытка. Фосфорная кислота образует с ионами трёхвалентного железа бесцветное комплексное соединение, что увеличивает контрастность изменения краски индикатора. Серная кислота применяется для создания нужного значения кислотности среды.
Рассчитаем тирование 100 мл 0,1 моль-экв. р-раFeSO4 р-ром KMnO4 в кислой среде ([Н+]=1моль/л).
31.Броматометрия: стандартные вещества и индикаторы. Броматометрическое определение сурьмы, фенола. КТхлороводородного раствора хлорида сурьмы (III).
Броматометрия является одним из методов редоксиметрии, в котором используется реакция окислениябромат-ионом BrO3-. В кислой среде брома-ионы восстанавливаются до бромида: BrO3-+6H++6 →Br-+3H2O.
Для ускорения реакции титрование ведут в нагретом и сильно кислом р-ре. При титровании первая избыточная капля KBrO3 вступает в реакцию с получившимися в р-ре бромид-ионами, выделяя свободный бром, который окрашивает р-р в бледно-желтый цвет: BrO3-. +5Br-+6H+→3Br2+3H2O.
Получаемая окраска слаба, и ТЭпо ней нельзя точно фиксировать.Некоторые орг. красители могут необратимоок-ся свободным бромом, и окрашенный ими р-р при этом обесцвечивается.В броматометрии чаще используют кислотно-основные индикаторы: метиловый оранжвы и метиловый красный. С необратимостью процесса приходится считаться при работе с этими индикаторами, т.к. окраска может исчезнуть до достижения ТЭ. Поэтому вконце титрования прибавляют еще несколько капель индикатора. При повторных же титрованиях индикатор вводят в р-р лишь после того, как прибавлен почти весь требуемый объем р-ра KBrO3.
Броматометрию применяют для определения мышья (III), сурьмы (III), таллия (I) и гидрозина в кислой среде, олова (II)и меди (I) в инертной атмосфере, многих других неорг. и орг. соединений (фенолов, аминов, аскорбиновой кислоты). Его можно применять не только для определения окислителей и восстановителей, но и для анализа орг. ненасыщенных, ароматических и гетероциклилических соединений.
Определение сурьмы основано на реакции: 3[SbCl6]3- +BrO3 - +6H+ →3[SbCl6]-+Br- +3H2O. Анализируемый р-р помещают в колбу на 100мл, доводят дистиллированной водой до метки. В колбу для титрован помещаюталиквоту исследуемого р-ра, нагревают до 600С, добавляют каплю метилового оранжевого и медленно титруют при перемешивании р-ром KBrO3 до обесцвечивания. Снова нагревают р-р, добавляют каплю индикатора, иесли р-р не обесцвечивается, продолжаюттитрование до исчезновения окраски.
Определение фенола. В анализируемый раствор вводится избыток бромат-бромидной смеси, которая в кислой среде выделяет свободный бром, реагирующий с фенолом: С6Н5ОН+3Br2 →С6Н2Br3ОН+3НBr, при добавлении к этому р-руKI непрореагировавший бром окисляет йодид до йода, который титруют стандартным р-ром Na2S203: Br2+2I- = 2Br-+I2
I2+ 2S2032- =2I-+ S4062-
В колбу для титрования помещают аликвоту (10 мл) исследуемого р-ра, прибавляют 12 мл бромат-бромидной смеси, 10 мл 1М р-ра серной кислоты, закрывают пробкой и оставляют на 30 мин, затем добавляют 1 г KI и снова закрывают пробкой. Через 5 мин титруют р-ром Na2S203, прибавляют в конце титрования, когдаокраска р-ра станет светло-жёлтой, крахмал, продолжают титрование до исчезновения синей окраски р-ра.
32 Йодометричопред меди (II). Окислительно-восстановительная способность пары I2/2I-. Kривая титрования йода Na2S203. Факторы, влияющие на ход КТ. Способы установления КТТ в йодометрии.
В основу йодометрических методов составляет полуреакция: [I3]- +2 =3I-. Нормальный потенциал окислительно-восстановительной системы [I3]- \ 3I-=0.545 В и занимает промежуточное положение между потенциалом сильных ок-ей и сильных вос-лей. Поэт йодометрические методы применяются при определении как ок-лей, так и вос-лей.
Йод, выделившийся в рез-те окисления I-, титруют Na2S203: 2 Na2S203+I2→ 2NaI+ Na2S406.
Свободный йод окрашивает р-р крахмала в синий цвет. Если к р-ру какого-нибудь вос-ля прибавить крахмал и титровать йодом, то после достижения ТЭ избыточная капля йода вызовет неисчезающую синюю окраску. Можно и наоборот, т.е. к р-ру йода в присутствии крахмала постепенно приливать вос-ль. В этом случае ТЭопределяют по обесцвечиванию синей окраски. Кроме крахмала применяют и др. индикаторы-хлороформ, бензол. По мере связывания йода во время титрования он переходит из неводной фазы, окраска бледнеет и, наконец, исчезает в ТЭ.
Йодометрию применяют для определенияок-лей: перманганатов, бихроматов,броматов, хлора, брома и др, а так же для определениявос-ей: сульфидов, сульфитов, тиосульфатов, орг. в-в. С помощью йодометрии возможно определение кислот. Метод основан на том, что реакция окисления йодидов йодатами происходит в кислой среде. Количество выделившегося йода при этом эквивалентно содержанию кислоты в р-р. Косвенно йодометрический метод анализа применяют также при определении ионов бария и свинца, осаждая их в виде хроматов с дальнейшим восстановлением хроматов йодидом калия.
Факторы, влияющие на вид КТ:
1) Количество . Чем больше принимает участие в ОВР, тем более пологая кривая будет наблюдаться.
2) Значение (разница) Е0. Чем больше ЭДС, тем больше скачок.
3) Температура. Повышение температуры увеличивает потенциал и окис-ля и вос-ля, поэтом КТ будет сдвинута по ОУ
4) Разбавление р-ра. Оно влияет, если стехиометрические коэффициенты не равны.
Дата добавления: 2016-01-05; просмотров: 77; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!