Синдром Шерешевского-Тернера (моносомия Х).
Кариотип 45,Х0. В клетках отсутствуют тельца полового хроматина. Частота 1:2000-1:5000. Синдром описали русский клиницист М.А. Шерешевський (1925) и Г.Тернер (1938).
Клинические диагностические признаки: женский фенотип; низкий рост, короткая шея с латеральными складками кожи (шея сфинкса), низкая граница роста волс на затылке, грудная клетка щитообразной формы с широко расставленными сосками, дисгенезия гонад, первичная аменорея, бесплодие.
Синдром Клайнфельтера.
Кариотип 47, ХХУ. Частота 1:400. Синдром диагностируется лишь у лиц мужского пола преимущественно при половом созревании.
Клинические признаки: высокий рост, длинные конечности, евнухоидизм, гинекомастия (увеличения молочных желез), отсутствие сперматогенеза, недоразвитие половых желез. Тельца полового хроматина оказываются в 80 % случаев. Иногда больные с синдромом Клайнфельтера имеют 48 и 49 хромосом (48, ХХXY; 49, ХХХХY). Чем больше Х-хромосом в кариотипе, тем высшая вероятность развития умственной отсталости.
Фенилкетонурия – наследственная болезнь аминокислотного обмена, обусловленная дефицитом фермента фенилаланин-гидроксилазы, который необходим для преобразования аминокислоты фенилаланина в тирозин. Это ведет к накоплению фенилаланина, фенилпировиноградной, фенилоцтовой и фенилмолочной кислот в крови, спинномозговой жидкости, тканях и их токсичного действия на ЦНС.
Локус (фенилгидроксилазы) расположен в длинном плече 12-й хромосомы.
Гемофилия служит классическим примером Х-сцепленного рецессивного аномального гена, который проявляется фенотипически у мужчин. У женщин-гетерозигот его действие подавляется доминантнымаллелем нормального свертывания крови. Отец-гемофилик никогда не передает ген гемофилии сыновьям. Поэтому его дети здоровые, но все дочери рождаются носителями болезни.
Дальтонизм является одной из наиболее распространенных аномалий, которые наследуются рецессивно, сцепленно с Х-хромосомой. Характеризуется нарушением восприятия красного и зеленого цветов. Принципы его наследования такие самые, как гемофилии.
2. Принципы классификация наследственных болезней.
Все наследственные болезни принято разделять на три большие группы: моногенные, болезни с наследственным предрасположением (мультифакториальные) и хромосомные.
Причиной развития моногенных болезней является поражение генетического материала на уровне молекулы ДНК, в результате чего повреждается только один ген. Сюда относятся большинство наследственных болезней обмена (фенилкетонурия, галактоземия, муковисцидоз, адреногенитальный синдром, гликогенозы, мукополисахаридозы и др.) моногенные болезни наследуются в соответствии с законами Менделя и по типу наследования могут быть разделены на аутосомно-доминантные, аутосомно-рецессивные и сцепленные с Х-хромосомой.
Болезни с наследственным предрасположением (мультифакториальные) являются полигенными, и для их проявления требуется влияние определенных факторов внешней среды. Общими признаками мультифакториальных заболеваний являются:
- Высокая частота среди населения;
- Выраженный клинический полиморфизм;
- Сходство клинических проявлений у пробанда и ближайших родственников;
- Возрастные отличия;
- Половые отличия;
- Различная терапевтическая эффективность;
- Несоответствие закономерностей наследования простым менделеевским моделям.
Хромосомные болезни могут быть обусловлены количественными аномалиями хромосом (геномные мутации), а также структурными аномалиями хромосом (хромосомные аберрации).
Наследственные болезни нельзя отждествлять с врожденными заболеваниями, с которыми ребенок рождается и которые могут проявляться сразу же после рождения или в более поздний период жизни. Врожденная патология может возникнуть в критические периоды эмбриогенеза под воздействием внешнесредовых тератогенных факторов (физических, химических и др.) и не передается по наследству.
Классификация наследственных заболеваний.
| Хромосомные | Моногенные | Мультифакториальные (полигенные) |
| А. Аномалии количества - половых хромосом: Синдром Шерешевского-Тернера, Клайнфельтера, синдром трисомии Х и др.; - аутосом: Болезнь Дауна, синдром Эдвардса, Патау и др. Б. Структурные аномалии хромосом: Синдром «кошачьего крика» и др. | А. Аутосомно-доминантные: Синдром Марфана, ахондроплазия, анемия Минковского-Шоффара, полидактилия и др. Б.Аутосомно-рецессивные: Фенилкетонурия, галактоземия, целиакия, муковисцидоз, адреногенитальный синдром, гликогенозы, мукополисахаридозы и др. В. Х-сцепленные рецессивные: гемофолия, миопатия Дюшена, ихтиоз и др. Г. Х-сцепленные доминантные: витамин Д-резистентный рахит, коричневая окраска эмали зубов и др. | А. ЦНС: эпилепсия, шизофрения и др. Б. Сердечно-сосудистые:ревматизм, атеросклероз и др. В. Кожные: атопический дерматит, псориаз и др. Г. Дыхательной системы:бронхиальная астма, аллергический альвеолит и др. Д. Выделительной системы: нефриты, мочекаменная болезнь, энурез и др. Е. Пищеварительной системы: язвенная болезнь, цирроз печени, неспецифический язвенный колит и др. |
https://studfiles.net/preview/6216626/page:4/
3. Принципы диагностики и лечения наследственных болезней.
Различают пренатальную и постнатальную диагностику. В пренатальной диагностике используют ряд методов, среди которых наиболее эффективным является амниоцентез (рис. 79).
Суть этого метода заключается в трансабдоминальном получении околоплодной жидкости и исследовании ее непосредственно путем микроскопии содержащихся в ней клеток или после культивирования последних. С помощью метода амниоцентеза возможно опреде-ление пола и кариотипа плода. Широкое распространение получил метод ультразвуковой диагностики (УЗИ).
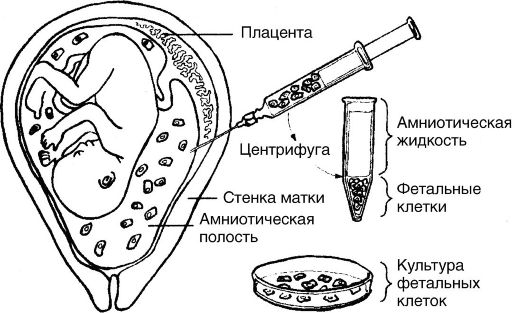
Рис. 79.Амниоцентез
Постнатальная диагностика наследственных болезней основывается на результатах клинического, параклинического и генетического обследования пациентов. Генетическое обследование основывается в первую очередь на результатах генеалогического анализа. В зависимости от показаний привлекают также цитогенетические, биохимические, иммунологические и другие методы.
Большое значение имеют методы массовой («просеивающей») диагностики с целью проверки населения на возможность скрытых форм наследственных аномалий.
Как и в случае других болезней, различают симптоматическое (воздействие на симптомы болезни), патогенетическое (воздействие на патогенез болезни) и этиологическое (воздействие на причину болезни) лечение наследственных болезней.
Симптоматическое и патогенетическое лечение наследственных болезней заключается в равных формах терапии. При некоторых наследственных болезнях прибегают к диетотерапии. Поскольку, например, патогенез такого наследственного заболевания, как галактоземия, связан с накоплением в клетках галактозы из-за отсутствия фермента ф-Д-галакто-1-фосфат-уридилтрансферазы, вследствие чего развиваются изменения в печени и головном мозге и, наконец, ослабление умственной деятельности, то лечение болезни обычно проводят исключением материнского молока и назначением диеты, не содержащей галактозы. Другой пример диетотерапии представляет лечение фенилкетонурии, которая может быть обнаружена простым исследованием мочи новорожденных. Новорожденному с этой болезнью назначается диета с пониженным содержанием фенилаланина в период раннего детства, что предупреждает умственную отсталость и другие симптомы фенилкетонурии.
С целью лечения часто прибегают к введению в организм недостающего фактора. Например, при пернициозной анемии, гемофилии и антигемофильной глобулиновой недостаточности прибегают к периодическим инъекциям недостающего в организме белка, что временно улучшает состояние больных. Для лечения ряда болезней используют переливание крови или удаление из организма токсических веществ с помощью лекарственных средств. Лекарственную терапию часто используют с целью индукции недостающих ферментов. Важное место в лечении наследственных болезней занимает хирургическое лечение путем удаления органов или частей орга- нов, коррекция повреждений или трансплантаций. Например, часто прибегают к удалению толстой кишки при полипозе, селезенки при сфероцитозе, почек при цистинозе.
Этиологическое лечение наследственных болезней, которое должно приводить к кардинальному исправлению наследственных аномалий, еще не разработано.
4. Медико-генетическая консультация: цели, задачи, методы работы.
Медико-генетическое консультирование-отрасль профилактической медицины, главной целью которой является предупреждение рождения детей с наследственной патологией. Современная генетическая консультация призвана служить интересам семьи и общества.
Цель: установления степени генетического риска в обследуемой семье и разъяснение супругам в доступной форме медико-генетического заключения.
Задачи медико-генетического консультирования:
1.про- и ретроспективное (до и после рождения) консультирование семей с наследственной или врожденной патологией
2.пренатальная диагностика врожденных и наследственных заболеваний
3.помощь врачам различных специальностей в постановке диагноза заболевания, если для этого требуются специальные генетические методы исследования
4.объяснение в доступной форме пациенту и его семье степени риска иметь больных детей и помощь им в принятии решения
5.ведение территориального регистра семей и больных с наследственной и врожденной патологией и их диспансерное наблюдение
6.пропаганда медико-генетических знаний среди населения.
Коротко говоря, задачей генетической консультации является составление генетического прогноза в семье индивидуума с аномалией физического, психического либо полового развития и выбор профилактических мероприятий по предупреждению рождения больного ребенка
5. Наследственные нервно-мышечные заболевания. Классификация, клиника и критерии диагноза.
Классификация ННМЗ
1. Первичные прогрессирующие мышечные дистрофии (первичные ПМД, миопатии)
2. Вторичные ПМД, или нейрогенные(вторичные) амиотрофии, т.е. спинальные и невральныеамиотрофии, обусловленные поражением на различных уровнях (тела или аксона) периферических двигательных нейронов
3. Врожденные непрогрессирующие миопатии
4. Миотонии
5. Пароксизмальные параличи
Общие признаки ННМЗ:
1.мышечная слабость проявляется симметрично и прогрессирует постепенно
2.мышечная слабость не сопровождается перманентной болью, хотя и возможны болезненные мышечные спазмы – крампи
3.при большинстве форм ПМД слабость раньше проявляется и преобладает в мышцах тазового или плечевого пояса и проксимальных отделах конечностей
4.сухожильные рефлексы снижаются пропорционально выраженности мышечной слабости
5. парестезии, расстройства поверхностной и глубокой чувствительности встречаются нечасто, обычно при невральныхамиотрофиях
6. заболевание, как правило, не влияет на функции тазовых органов
Некоторые симптомы и феномены, характерные для ПМД
1. Миопатическая или утиная походка
2. Феномен Тренделенбурга: у больного стоящему на одной ноге таз на стороне поднятой ноги опускается, а не поднимается, как это происходит в норме
3. Феномен Дюшена: при ходьбе таз опускается в сторону неопорной ноги, а туловище при этом отклоняется в противоположную сторону
4. «Икры гнома»: псевдогипертрофия икроножных мышц в связи с их жировой инфильтрацией и разрастанием в них соединительной ткани
5. Симптом «осиной талии»: своеобразная перетяжка туловища в связи с атрофией косых и прямых мышц живота, при сохранности его поперечных мышц
6. «Лягушачий живот»: низкий тонус и гипотрофия мышц живота, лежа он распластан, стоя - выступает вперед
7. Симптом Бивора: выстояние пупка
8. Тест «встряхивания»: разболтанность, избыточный объем движений в суставах…
9. Лицо «Сфинкса»: миопатическое лицо, гипомимия…
10. Губы «тапира»: атрофия круговой мышцы рта-губы выпячены вывернуты
11. Улыбка «Джоконды»6 поперечная улыбка
12. Симптом Зинченко: больному особенно трудно подниматься по лестнице…
13. Симптом Шерешевского – Говерса: больной из положения лежа встает через ряд последовательных промежуточных движений + с взбиранием руками по своему телу
14. Пяточно-ягодичная проба
15. Симптом Оршанского: Переразгибание в коленном суставе лежа (гипермобильность)
6. Миопатия Дюшена, Беккера, Ландузи–Дежерина. Клиника, диагностика, дифференциальная диагностика, медико-генетические аспекты.
Миодистрофияпсевдогипертрофическая ДЮШЕННА (Х-сцепл. рецессивн. тип)
утрата мышечного белка дистрофина
наиболее злокачественная форма первичных миодистрофий
дебют в раннем детском возрасте
с 2-5 лет уже развивается слабость мышц ТАЗОВОГО пояса, бедер, утиная походка
распространение процесса восходящее (до плечевого пояса)
особенно типична псевдогипертрофия икроножных мышц
со временем возникает слабость мышц лица, языка, глотки, гортани, дыхательных мышц
возможны сухожильные ретракции (чаще пяточного)
развиваетсякардиопатия
возможен адипозо-генитальный синдром, гипоплазия надпочечников, остеопороз
у 30% отставание в интеллектуальном развитии
высокая степень гиперферментемии (КФК)
ПоздняяпсевдогипертрофическаяМиодистрофия БЕККЕРА-КИНЕРА (Х-сцепл. рецессивн. тип)
дебют от 5 до 20 лет чаще 10-15 лет
течение медленно прогрессирующее
распространение мышечных дистрофий как при миодистрофииДюшена
поражение сердца выражены меньше
доживают до 30-60 лет, могут иметь детей, интеллект сохранен
повышение активности КФК умеренное
называют мягкой формой миодистрофииДюшена
качественное изменение белка дистрофина
Плечелопаточно-лицевая миодистрофия ЛАНДУЗИ-ДЕЖЕРИНА (по аутосомно-рецессивному типу)
дебют чаще к 20 годам, иногда несколько позже
слабость и гипотрофия мышц лица, особенно круговых глаз и рта, мышц плечевого пояса
рано губы тапира, лицо сфинкса, улыбка Джоконды, крыловидные лопатки
далее: слабость передней зубчатой, большой грудной, нижних отделов трапецивидных мышц, широчайшей мышцы спины, двуглавой, трехглавой мышц
далее: слабость перонеальных мышц (появляется степаж)
далее: в меньшей степени проксимальные мышцы нижних конечностей
возможна умеренная псевдогипертрофия икроножных и дельтовидных мышц
сухожильные рефлексы постепенно снижаются
интеллект сохранен
течение относительно мягкое
гиперферментемия умеренная
женщины в 3 раз чаще мужчин болеют
Дата добавления: 2018-08-06; просмотров: 311; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!