Некоторых углеводородов при разных температурах
(данные Россини и др.)
| 800 |
Углеводород
298,16
Температура, К
500
Метан...................
Этан........................
Пропан ...............
Бутан.......................
Изобутан................
Пентан.................
Гексан ...................
Гептан .................
Октан......................
Декан ................
Тетрадекан ...
Этилен.................
Пропилен ....
Бутен-1 ...........
Изобутилен . . . Пентен-1 ....
Гексен-1..................
Гептен-1 .............
Октен-1 ...........
Децен-1 .............
Цнклопентан . . . Циклогексан . . . Метилциклопентан
Бензол ...................
Толуол ................ .
Кумол .......
Ацетилен.................
Бутадиен-1,3 . . .
-50,828
-32,898
-23,498
-17,143
-20,928
-8,371
-0,293
8,119
16,532
33,358
66,968
68,146
62,741
71,531
58,095
79,357
87,644
96,057
104,468
121,292
38,633
31,767
35,786
129,900 ■
122,343
137,028
209,280
150,722
-32,817
4,889
34,447
60,857
60,229
90,031
118,446
147,076
175,874
233,507
369,618
80,546
93,965
123,682
112,085
151,601
180,315
209,108
215,439
295,533
122,426
142,603
138,120
164,243
175,004
232,125
197,540
180 189
-2,302 66,633 127,443 185,039 188,852 245,983 305,875 366,024 426,503 547,468 789,423 102,501 145,700-206,974 198,771 266,118 326,554 387,037 447,514 568,472 256,994 317,928 ЗОЬСбЗ 221,157 260,^88 382,889 180.733 228,029
41,039 151,563 255,362 355,096 364,810 458,857 561,322 664,072 767,331 873,802 1386,773 134,317 218,278 321,197 318,351 422,443 525,743 628,992 732,256 938,722 441,279 554,918 423,071 300,476 378,793 588,900 159,423 294,236
Таким образом, при 7'>305СС реакция распада гептана становится более термодинамически вероятной.
Подсчитаем теперь значения КР для разных температур из выражения
lgtfp = -АО/19,137-
и сведем все данные
■ Т, К............................... ............ 298 578 800
7" °с.............................. ............. 25 305 527
|
|
|
ДО, кДж/моль '.'.'.'.'. 39,904 0 -31,607
IgKp.................................. .......... —6,99 0 2,06
АГр...................................... ........ 1,2- 10"' 1 114,8
Константа равновесия для этой реакции имеет вид:
д. _ Рс4н8Рс3н8
170
Принимая за х мольную долю гептана, прореагировавшего К моменту установления равновесия (AG = 0), выражаем парциальные давления через мольные доли
РС.Ня — Рс3Н8 —
\+х'
Рс7н6:
1 + *
так как в смеси имеется х молей бутена, х молей пропана и 1 — х молей гептана, а всего 1 -+- х.
Подставляя эти значения в выражение для Кр, находим х, т. е. парциальные давления пропана, бутана и гептана, или, что то же самое, их равновесные концентрации при разных температурах. Получаем:
| 527 |
Температура, °С.......................... 25 304
Концентрация, %
| 49,92 49,92 0,16 |
С3На....................................... 0,011 41,0
С4Н3....................................... 0,011 41,0
С7Н,6............................... ■ . . 99,978 18
|
|
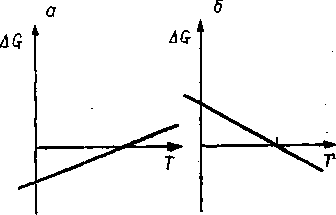
| Рис. 37, Зависимость изменения энергии Гиббса от температуры для экзотермических (а) и эндотермических (б) реакций. |
В разобранном примере мы имели дело с эндотермической реакцией распада, которая идет с поглощением тепла. Обобщая, можно сделать заключение, что для всех эндотермических реакций при увеличении температуры сверх определенного предела (при ДО = 0) равновесие реакции смещается слева направо, т. е. в сторону образования продуктов реакции. Такие реакции можно назвать принципиально высокотемпературными (рис. 37,6). Наоборот, для большинства реакций синтеза, таких, как гидрирование, алкилирование, полимеризация, являющихся экзотермическими, имеет место обратная картина, и они могут быть названы принципиально низкотемпературными. При падении температуры ниже определенного предела (при AG = 0) их термодинамическая вероятность растет (рис. 37,а).
|
|
|
Для реакций, идущих с увеличением или уменьшением объема, на состояние равновесия оказывает влияние не только температура, но и давление. Приближенно зависимость AG от давления Р выражается уравнением:
ДО = ДОо± 19,13\gPT
где AGq — изменение энергии Гиббса при давлении 101325 Па.
Качественно этот вопрос решается, исходя из принципа Ле-Шателье. Повышение давления способствует реакциям гидрирования, алкилирования, полимеризации и другим, идущим
171
,с уменьшением объема. Наоборот, для реакций, протекающих с увеличением объема (распад, дегидрирование и др.), термодинамическая вероятность увеличивается с понижением давления. Для реакции изомеризации и других, протекающих без изменения объема, давление не оказывает влияния на величину AG и Кр.
|
|
|
Итак, термодинамический анализ позволяет выяснить направление обратимой реакции при различных условиях, дает возможность подсчитать теоретические равновесные концентрации продуктов реакции, исходя из константы равновесия. Однако для практического осуществления реакции одной ее термодинамической вероятности недостаточно. Надо, чтобы она протекала и с приемлемой для технологических целен скоростью и за время процесса достигалась необходимая степень превращения.
Степень превращения можно выразить через концентрации исходного вещества в начальный период реакции С0 и к моменту времени т — С:
с0-с с„
Большинство реакций углеводородов нефти характеризуется небольшими скоростями и соответственно значительными энергиями активации. Для увеличения скорости реакции приходится повышать температуру или применять катализаторы. Как известно, для большинства реакций при повышении температуры на 10 °С скорость реакции увеличивается в 2—4 раза. Повышением температуры и достигается требуемая степень превращения исходного сырья. На практике часто бывают случаи, когда даже в ущерб термодинамической вероятности приходится повышать температуру, с тем чтобы получить реальный выход продуктов реакции хотя бы и с малыми равновесными концентрациями, т. е. с небольшим значением Кр для данной реакции.
|
|
|
Давление не влияет на скорость мономолекулярных реакций (распад, изомеризация, дегидрирование), поскольку каждая молекула в них реагирует самостоятельно.
Для бимолекулярных реакций синтеза (полимеризация, гидрирование, алкилирование) повышение давления увеличивает возможность столкновения реагирующих молекул и поэтому увеличивает скорость реакции.
Реакции, происходящие при термическом крекинге, в основном подчиняются уравнениям первого порядка, для которых константа скорости k определяется через концентрации Со и С и время реакции т:
Энергия активации реакций крекинга, определяющая их скорости, колеблется в пределах 200—280 кДж/моль.
Преобладающим типом химических превращений при деструктивных процессах переработки нефти является распад углеводородов. Термическая стабильность углеводородов неодинакова и
172
зависит как от их молекулярной массы, так и химического строения и тесно увязана с энергией связей между атомами в молекулах.
Энергией связи называется такое количество энергии, которое необходимо затратить для разрыва (диссоциации) определенного типа связи между атомами. При возникновении связи, т. е. при соединении атомов в молекулы, происходит переход системы в более устойчивое состояние, сопровождающееся выделением тепла. Энергия образования связи, следовательно, положительна. При распаде молекул тепло поглощается и энергия разрыва связи отрицательна.
Ниже приводятся усредненные данные по энергиям связи (в кДж/моль) между атомами углерода и углерода с водородом в углеводородах различных классов:
Н—Н................................. ...... 436 С=С.................................... ...... 610
С,ЛИф-СалИф •■•■■ 2^8 С=С ...... 836
СалИф-Сар........................... ...... 332 Салиф--Н.............................. 392
Сар—Сар............................. ...... 407 Сар-Н................................. ...... 426
Однако следует иметь в виду, что энергии связи С—С и С—Н в молекулах не однозначны. На их значение влияет строение молекулы и местоположение данной связи в молекуле. Опытные данные, например, показывают, что отрыв водорода легче всего
происходит от третичного атома углерода — СН, несколько труднее от вторичного уСН2> еще труднее от первичного —СН3.
Наименьшая вероятность отрыва водорода у метана СН4. В ряду углеводородов СпН2П+2 связь С—С значительно ослабляется по мере увеличения числа углеродных атомов в молекуле.
Основные типы углеводородов нефти по термической стабильности в общем виде располагаются в следующий возрастающий ряд: алканы, нафтены, ароматические, нафтено-ароматические, полициклические ароматические.
§ 36. МЕХАНИЗМ ТЕРМИЧЕСКИХ ПРЕВРАЩЕНИЙ УГЛЕВОДОРОДОВ
Термические превращения нефтяных фракций — весьма сложный химический процесс. Сырье состоит из большого числа индивидуальных компонентов. Совершенно очевидно, что предсказать или проследить судьбу каждого компонента сырья при воздействии высоких температур невозможно. Поэтому на практике о результатах того или иного высокотемпературного процесса судят обычно по выходам целевых продуктов: газа, бензина, кокса, а также по групповому или, в лучшем случае, компонентному составу бензинов. Однако многочисленные исследования термических превращений отдельных углеводородов позволяют делать
173
выводы о характерных для данного класса углеводородов типах реакций.
Распад алканов.Термодинамические расчеты показывают, что углеводороды этого ряда, начиная с пентана, при повышенных температурах склонны лишь к реакции распада по связи С—С, с образованием алкена и алкана:
С„Но„+2 —>■ CmH2m + С^Но^+о
Полученные предельные осколки вновь распадаются на алкен и алкан. Низкомолекулярные углеводороды—этан, пропан и бу-таны — могут также дегидрироваться:
СИН;п + 2 *" СпНг/1 + Но
В результате в газах накапливается водород. Распад алканов может происходить по всем связям С—С. Место разрыва, а следовательно, преимущественное образование тех или иных продуктов реакции зависят от температуры и давления. Чем выше температура и ниже давление, тем место разрыва углеродной цепи все больше смещается к ее концу и значительно возрастает выход газообразных продуктов. При температуре около 450°С разрыв происходит посередине цепи. Повышение давления также сдвигает место разрыва к центру молекулы. Поэтому крекинг под давлением позволяет получать больше целевых жидких продуктов.
В ряду алканов метан является наиболее термически устойчивым, так как в нем отсутствуют связи С—С. Разложение метана на углерод и водород возможно при очень высоких температурах (примерно 1500°С). Распад этана, пропана и бутанов изучали многие исследователи. Для этана характерна реакция дегидрирования:
700 °С
СоН6 *- С2Н4 + Н2
Пропан примерно в равной степени реагирует по двум направлениям
600 °С |--- > С2Н1+СН4
С3Н8
1—► С3Н, + Н2
Бутан при 500—600 °С распадается по всем связям С—С и в незначительной степени дегидрируется:
60%
—»■ сн4 + с3н6
30%
| С4Н1. |
—>- с2н6 + с2н4
10%
----- •> С4Н8 + Но
Изобутан, наоборот, при этих температурах в основном дегидрируется:
70 %
----- *■ Н2 + изо-С4Н8
| цэо-С4Н|о |
28?»
----- > СН4 + СоНа
2%
------- >■ C2H<j + С2Н4
174
Для углеводородов с числом углеродных атомов более четырех при обычных температурах крекинга распад происходит в центре углеродной цепи.
Написанные выше схемы химических реакций распада алканов не раскрывают еще всей сложности механизма процесса образования водорода, предельных и непредельных осколков. Большинство ученых считает, что распад алканов имеет.цепной характер и подчиняется теории свободных радикалов, выдвинутой Ф. О. Рай-сом и С. К- Райсом. Кратко эта теория сводится к следующему.
1. Так как энергия диссоциации связи С—С меньше, чем связи
С—Н, то первичный распад под воздействием повышенной темпе
ратуры происходит по связи С—С. В результате образуются ра
дикалы различной молекулярной массы, например:
СНз(СН2)5СН3 —-> С4Н9 • + С3Н7 •
2. Доказано, что при температуре около 600 °С и при атмосферном или пониженном давлении радикалы метил(СН3«) и этил(С2Н5«) способны к кратковременному самостоятельному существованию. При повышенных давлениях этой способностью обладают радикалы и большей молекулярной массы. Ненасыщенные вещества со свободной валентной связью, не распадающиеся мгновенно на более устойчивые соединения, называются свободными радикалами. К ним относится также и водородный радикал Н«.
3. Радикалы более высокой молекулярной массы, не обладающие в данных условиях хотя бы минимальной стабильностью, мгновенно снова распадаются с образованием устойчивого этиленового углеводорода и нового радикала, в том числе и водородного атома, например:
I—> С4Н8 + СНз • —С5Нц —
>—-> С5Н10+Н.
При этом, как видно, концентрация свободных радикалов в реакционной смеси повышается.
4. Свободные радикалы, сталкиваясь с молекулами исходного
сырья, порождают цепную реакцию образования новых радикалов:
r'H + Н • —► На ■+■ R' R'H + R • —» RH + R' • [
Единственным результатом- соударения свободного радикала с молекулой сырья будет отрыв водорода от предельного углеводорода. С разной степенью вероятности этот отрыв будет происходить от третичных, вторичных и первичных атомов углерода, порождая радикалы различного строения, например при пиролизе пропана:
-> сн:,сснсн3 + сн4
СН3СН2СН3 + СНз
—> СН;<СН2СН2—+ СН4
175
При 600 °С вероятность реакции свободных радикалов с водородом по месту вторичных и первичных атомов углерода находится в соотношении 1 :2. Время жизни свободных радикалов очень мало, порядка 10_3-^10~4 с, так как :ia это время они успевают встретиться с неразложенными молекулами сырья. Энергия активации этого типа реакций 40—80 кДж/моль, т. е. значительно ниже, чем для прямого распада алкана. Свободные радикалы при развитии цепной реакции вновь и вновь возникают, и, таким образом, очень большая часть исходных молекул алкана вовлекается в реакцию. Следовательно, основная масса продуктов реакции получается в результате развития именно цепной реакции через свободные радикалы, а не в результате прямого разрыва углеродной цепи. Эта реакция служит только началом длинного ряда превращений, как бы первичным толчком.
5. При достижении равновесия вероятность встречи свободных радикалов друг с другом становится не меньшей, чем вероятность их соударения с молекулами исходного сырья. Это приводит к обрыву цепи, так как при взаимодействии двух свободных радикалов образуются стабильные низкомолекулярные алканы или молекула водорода:
R. + R —> RR' R • + Н • —* RH Н • + Н • —> Н2
В конечном итоге в продуктах реакнии накапливаются устойчивые при данной температуре предельные и непредельные углеводороды, а также молекулярный водород.
Термические превращения непредельных углеводородов. В сырье для крекинга ненасыщенные углеводороды отсутствуют, но роль их в химии крекинга очень велика, так как они всегда образуются при распаде углеводородов других классов. Для непредельных углеводородов характерно большое разнообразие химических превращений. На примере олефинов особенно легко проследить решающее влияние температуры на направление превращения. Низкие температуры и высокие давления стимулируют реакции уплотнения низкомолекулярных олефинов
мСпН2п —> (СпНш)п
Чем выше давление, тем глубже идет полимеризация. Однако при повышении температуры термодинамическая вероятность полимеризации резко падает и равновесие смещается в обратную сторону. Чем больше время пребывания сырья в зоне высоких температур, тем глубже идет распад продуктов уплотнения. В обычных условиях крекинга (около 500 °С, давление до 7 МПа) олефины, образовавшиеся при распаде предельных компонентов сырья или в результате .уплотнения в начальный период крекинга низкомолекулярных алкенов, в основном претерпевают распад. Разложение
176 •
2
олефинов может протекать в различных направлениях:
CrtHtn —>• 2CmH2OT (деполимеризация)
С„Н,„ —у CmH2m + С,Н2(7 (распад)
С„Но„ —> СтЬит-2 + 0^2^+2 (распад)
С«Нг„ —> CmH2m+2 + C^H^-f СрН2р-2 (деструктивная конденсация)
СПН:2Я —> СотНпт-2 + Н2 (деструктивная конденсация)
Анализ энергетических особенностей молекулы олефина показывает, что наименьшей энергией диссоциации обладает связь С—С, находящаяся в р-положении по отношению к двойной связи:
-с-
-с—с=с—с—с—с-
Y p a a P Y в
Это так называемое правило $-связи определяет наиболее вероятное место распада в углеродной цепи. Заметим тут же, что в различных реакциях отрыв водорода от атома углерода также идет легче от р-углерода.
Термическая устойчивость высших олефинов близка к устойчивости алканов.
Механизм распада алкенов, так же как и алканов, имеет цепной характер. Первичный распад по р-связи дает начало двум радикалам. Так, для пентена:
СНз—CH2-fcH2—СН=СН2 -^ СНз—СН2 • + • СН2-СН=СН,
Далее
C5H,0 + R —► RH + СН3СН2СНСН=СН2
Радикал CsHg* весьма неустойчив и сразу распадается на бутадиен и радикал метил:
СН3СН2СНСН=СН2 —> СН2=СНСН=СН2 + CHj.
I
Дальнейшее развитие цепной реакции идет через радикал метил. Этот механизм хорошо объясняет получение из пентена бутадиена и метана:
С5Н10 —*■ С4Н6 + CHi
При более высоких температурах (600—700°С) аналогичные реакции осуществляются и для низших олефинов С2—Q- Так, из этилена получается бутадиен и водород. Эта реакция характерна для пиролиза:
СН2=СН2 — у СН2=СН. + Н.
177
Связь С = С прочнее связи С—Н, поэтому первичный распад приводит к образованию радикала винила и водородного атома. Свободные радикалы реагируют с этиленом:
сн2=сн2 + сн2=сн • —> сн:;=сн. + сн2=сн2 сн2=сн2 + н • —> н2 + сн2=сн •
Обе реакции снова приводят к образованию винила, и естественно, что реакция заканчивается в результате их соударения:
сн2=сн • + • сн=сн2 —> сн2=снсн=сна
Суммарно реакция выражается уравнением: 2С2Н4 ^ > С<Нб + Н2
Условия пиролиза (температура до 700°С и низкое давление) способствуют также протеканию реакций дегидрирования алкенов:
С(Н8 —*■ С(Нв + Н2
Итак, в продуктах пиролиза накапливаются диолефины. Эти углеводороды весьма склонны при температурах около 700°С к уплотнению путем полимеризации (1) и конденсации с алкенами (2) с образованием циклических углеводородов:
2СН2=СНСН=СН2
_^>^^/СН=СН2
винилцнклогексен
СН2
//
сн сн2
L +U -*■ i^i- ■ (2)
\
сн2
Получающиеся при конденсации циклоолефины легко дегидрируются до ароматических углеводородов. Этот так называемый диеновый синтез является одним из главных путей образования ароматических углеводородов при пиролизе.
Крекинг нафтенов.Для нафтеновых углеводородов наиболее характерны следующие типы превращения при высоких температурах:
1) деалкилирование или укорочение боковых парафиновых цепей;
2) дегидрирование кольца с образованием циклоолефинов и ароматических углеводородов;
178
3) частичная или полная дециклизация полициклических наф-тенов после деалкилирования;
4) распад моноциклических нафтенов на олефины или парафин— диолефин.
Деалкилирование— реакция, аналогичная распаду алканов. Термическая устойчивость боковых парафиновых цепей значительно ниже устойчивости кольца. Поэтому расщепление цепей является преимущественным направлением первичного распада ал-килнафтенов. При температуре около 500°С расщепление происходит главным образом посередине цепи. Деалкилирование — типичный пример последовательного типа реакций. По мере увеличения продолжительности нагрева боковые цепи становятся короче. Труднее всего отщепляется метальная и этильная группы. Полное деалкилирование возможно лишь при более высоких температурах. Повышение давления препятствует деалкилированию. Укороченная боковая цепь, так же как и отщепленный осколок, могут быть либо насыщенными, либо ненасыщенными. При исчерпывающем деалки-лировании циклический радикал насыщается водородом, всегда присутствующим в продуктах распада.
Дегидрирование — более высокотемпературная реакция, ведущая к накоплению в продуктах крекинга и пиролиза циклооле-финов и ароматических углеводородов. Реакции благоприятствует пониженное давление:
-н* l^JI -н2" li^J)
О-
-н2 Ц II —н2
Бициклические нафтены при этой реакции могут дать начало углеводородам рядов тетралина и нафталина. При пиролизе дегидрирование шестичленных нафтенов наряду с диеновым синтезом является наиболее вероятным путем глубокой ароматизации сырья.
Дециклизация полициклических нафтенов приводит к последовательному упрощению молекул и сопровождается деалкилирова-нием. Схематично эти превращения можно представить в следующем виде:
|
|
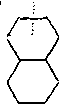
RwR R'wR"
распад I\ деалкилирование /\
+ С^Нгц + г + С„Н2П
Одновременно (особенно при высоких температурах) протекают реакции дегидрирования. Циклопентан, циклогексан и их
179
ближайшие гомологи при 550—600°С распадаются на два непредельных осколка:
|
|
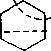
С2Н4+ ^Нд
2C3HG
Распад не имеет цепного характера. Термодинамически и кинетически реакция распада конкурирует с реакцией дегидрирования. Поэтому в продуктах крекинга можно обнаружить в сравнимых количествах и непредельные, и ароматические углеводороды. При 700—800°С циклогексан распадается иначе, с образованием бутадиена
СбНи —> СэНб + С[Нб
Крекинг ароматических углеводородов.Ароматические углеводороды наиболее термически устойчивы. Поэтому они накапливаются в жидких продуктах крекинга тем в больших количествах, чем выше температура процесса. При пиролизе ароматические углеводороды являются главной составной частью так называемой смолы пиролиза.
Ароматические углеводороды с длинными боковыми цепями способны деалкилироваться. Преимущественное место отрыва боковой цепи находится между первым и вторым атомами углерода в цепи, т. е. в р-положении, от углерода кольца. Поэтому при деалкилировании получаются главным образом монометилирован-ные ароматические углеводороды.
Голоядерные углеводороды любой степени цикличности (бензол, нафталин, антрацен и др.), так же как и алкилированные углеводороды с короткими боковыми цепями, практически не подвергаются распаду. Единственным направлением их превращений является конденсация с выделением водорода. В результате происходит накопление полициклических углеводородов. В результате конденсации бензола, нафталина и других голоядерных углеводородов образуются дифенил, динафтил и им подобные углеводороды:
2СбНе —*■ СаН5—СвН:; + Нг
2СюН8 —> С10Н7—С10Н7 + Н2
Эти реакции, согласно воззрениям Тиличеева, также легко объясняются радикально-цепным механизмом. Образующийся радикал водорода Н* порождает ароматические радикалы
с6нв + н« —> н2 + свн5-
которые в дальнейшем рекомбинируют, что и приводит к образованию конденсированных молекул.
Развитие реакций конденсации разнообразных циклических углеводородов приводит в конечном итоге к образованию карбои-
180
дов (кокса). Постепенное увеличение молекулярной массы, повышение содержания углерода и потерю водорода в результате конденсации ароматических структур можно изобразить следующими схемами:
|
|
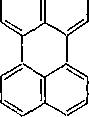
|
|
 -н2
-н2
-н2
(3)
нафталин
динафтил
перилен
|
|
|
|
|
|
-Н2
-Н2
(4)
антрацен
диантрацил
фенантроперилен
| \ |
| сн2 |
СН2—СН2
| 3\ |
сн
:сн2
СН2—СНг^
дибутилбензол
-На
фенантрен
(5)
Эти и аналогичные им реакции по мере углубления и приводят к образованию твердых карбоидов, содержащих минимальное количество водорода.
Итак, мы видим, что нефтяной кокс не является модификацией углерода, получающейся при распаде углеводородов на элементы, а имеет углеводородное строение. Если целью процесса не является получение нефтяного кокса, то коксообразование весьма нежелательно.
Что касается сернистых соединений, присутствующих в сырье для крекинга и пиролиза, то они либо разлагаются с выделением сероводорода, меркаптанов и углеводородных осколков (см. § 7), либо, благодаря своей термической устойчивости (тиофены и им подобные), накапливаются в более высокомолекулярных продуктах.
Подводя итог рассмотрению химических превращений углеводородов различного строения при температурах крекинга и пиролиза, можно сделать вывод, что при деструктивной переработке нефтяного сырья должны осуществляться следующие основные реакции: распад, деалкилирование, дегидрирование, полимеризация, циклизация непредельных, дециклизация нафтенов,
181
деструктивная конденсация олефинов, конденсация моноолефинов в диолефины, конденсации ароматических углеводородов, реакции глубокого уплотнения до кокса. От глубины этих реакций и преобладания того или иного типа превращений углеводородов сырья и зависят выходы газа, бензина, промежуточных фракций, крекинг-остатка и кокса, а также качество и химический состав целевых продуктов.
В свою очередь, степень превращения сырья и развитие перечисленных химических процессов зависят и от природы сырья и от параметров процесса.
§ 37. ВЛИЯНИЕ РАЗЛИЧНЫХ ФАКТОРОВ НА ПРОЦЕСС ТЕРМИЧЕСКОГО КРЕКИНГА
При любом термическом процессе в результате переработки образуются газы, бензин, средние дистиллятные фракции (керосино-газойлевые), тяжелые остаточные фракции и кокс. Выход, соотношение между продуктами реакции, свойства этих продуктов зависят от многих факторов, но главную роль играют состав сырья, температура, давление и продолжительность реакции.
Параметры процесса. Состав сырья. В одинаковых условиях крекинга скорость реакции растет с повышением температуры кипения сырья. Такая особенность объясняется различной термической стабильностью углеводородов. Высокомолекулярные парафиновые углеводороды, а также ароматические углеводороды с длинной боковой парафиновой цепью менее термически стабильны, чем низкомолекулярные углеводороды. Поэтому при крекинге последних будет образовываться меньше продуктов разложения.
Если термический крекинг ведут'при умеренных температурах, то заметного изменения структуры молекул расщепляющихся углеводородов не происходит. Так, при крекинге твердого парафина в основном образуются парафиновые и олефиновые углеводороды нормального строения, а при крекинге газойлей с высоким содержанием циклических углеводородов — бензиновые фракции, имеющие преимущественно нафтеновый и ароматическ"й характер.
Температура и продолжительность процесса. Эти факторы, влияющие на выход и качество продуктов термического крекинга, при определенных температурах взаимозаменяемы. Увеличивая температуру крекинга и уменьшая продолжительность времени пребывания в зоне высоких температур, можно получить ту же глубину разложения сырья, что и при более мягкой температуре, но большей длительности крекинга.
Термическое разложение углеводородов начинается при 380— 400°С. С увеличением температуры скорость крекинга быстро растет. Повышение температуры крекинга при постоянном давлении и постоянной степени превращения приводит к повышению содержания легких компонентов, к снижению выхода тяжелых фракций и кокса. Выход газа при повышении температуры заметно
182
увеличивается, причем растет содержание в нем непредельных углеводородов. t
Давление. При увеличении давления повышаются температуры кипения сырья и продуктов крекинга. Поэтому изменением давления можно влиять на фазовое состояние в зоне крекинга. Термический крекинг может осуществляться в паровой, жидкой и смешанной фазах.
^" В паровой фазе обычно проводится крекинг бензина, керосино-газойлевых фракций, для которых температура крекинга выше критической. Повышение давления при парофазном крекинге уменьшает объем паров сырья и продуктов крекинга, а это позволяет увеличить производительность установки или повысить продолжительность пребывания сырья в зоне реакции.
При парофазном крекинге давление существенно влияет на состав продуктов крекинга, поскольку благодаря давлению увеличивается скорость протекания вторичных реакций — полимеризации и гидрирования непредельных углеводородов, конденсации ароматических углеводородов и ряда других. При этом уменьшается выход газа.
Влияние давления на жидкофазный крекинг тяжелых видов сырья (мазута, гудрона) невелико. При смешаннофазном крекинге давление способствует гомогенизации сырья — газ частично растворяется в жидкости, уменьшая ее плотность, а сама газовая фаза уплотняется. Такое изменение фазовых условий увеличивает время пребывания в зоне крекинга и приводит к уменьшению потерь виде газа.
Тепловой эффект реакции крекинга.При термическом крекинге одновременно протекают реакции термического распада и реакции уплотнения. Первые из этих реакций идут с поглощением теплоты, вторые — с выделением теплоты.
Суммарный тепловой эффект процесса деструктивного разложения зависит от того, какие из этих реакций преобладают. Суммарный тепловой эффект термического крекинга отрицателен, и поэтому необходимо подводить тепло со стороны.
Теплота реакции термического крекинга выражается в расчете на 1 кг сырья или на 1 кг образующегося бензина. Она составляет 1,26—1,37 МДж/кг бензина при крекинге газойля и 0,63— 0,75 МДж/кг бензина при крекинге мазута.
Коксообразование и газообразование при крекинге. Врезультате сложных реакций полимеризации и конденсации из непредельных и ароматических углеводородов образуется твердый углеродистый остаток — кокс. Образование кокса при термическом крекинге — нежелательное явление, так как оно влияет на продолжительность безостановочного пробега установок. Из-за на копления кокса в змеевиках печей установки термического крекинга приходится часто останавливать на выжиг кокса.
Коксообразование зависит от свойств сырья крекинга и от таких параметров процесса, как температура и время пребывания в зоне реакции. Образование кокса ограничивает глубину крекинга
183
для тяжелых и средних видов сырья. С повышением температуры и времени пребывания сырья в зоне реакции выход кокса растет.
При крекинге легких видов сырья продуктов уплотнения образуется немного, и на выход целевого продукта — бензина главным образом влияет газообразование. Выход газа при крекинге сначала пропорционален выходу бензина, по мере углубления процесса относительный выход газа увеличивается. При очень глубоком крекинге скорость образования газа из бензина начинает превышать скорость образования бензина.
Коксообразование и газообразование не позволяют достичь мак^ симального выхода бензина на промышленных установках, и поэтому часть крекируемого сырья остается непревращенной. По некоторым данным выход бензина при крекинге тяжелого дистил-лятного сырья не превышает 50% от максимально возможного выхода.
Непревращенное сырье для увеличения выхода целевого продукта возвращается на повторный крекинг, который затем про,во-дится в самостоятельной аппаратуре, отдельно от свежего сырья (крекинг гуськом) или в смеси со свежим сырьем (крекинг с рециркуляцией).
§ 38. ПРОДУКТЫ, ТЕХНОЛОГИЧЕСКАЯ СХЕМА, РЕЖИМ И МАТЕРИАЛЬНЫЙ БАЛАНС ТЕРМИЧЕСКОГО КРЕКИНГА
Продукты термического крекинга. При термическом крекинге получают газ, бензин, газойль и крекинг-остаток.
В газе термического крекинга содержатся предельные (от метана до бутана) и непредельные (от этилена до бутиленов) углеводороды, водород и сероводород. Соотношение компонентов газа зависит от температуры и давления процесса. Состав газа термического крекинга приведен в табл. 15 (см. гл. VIII). Газ термического крекинга направляется для дальнейшей переработки на газо-фракционирующую установку.
Бензины термического крекинга по углеводородному составу отличаются от прямогонных. Если в прямогонных бензинах в основном содержатся парафиновые и нафтеновые углеводороды, то в крекинг-бензине много непредельных и ароматических углеводородов. Октановое число крекинг-бензинов составляет 66—68 пунктов по моторному методу в чистом виде. По своей антидетонационной стойкости крекинг-бензин не соответствует требованиям, предъявляемым современными автомобильными двигателями.
Кроме того, кр.екинг-бензины обладают низкой химической стабильностью. Содержащиеся в крекинг-бензинах непредельные углеводороды, в особенности те, в молекуле которых имеются две" двойных связи, под воздействием света, тепла и растворенного кислорода конденсируются, полимеризуются, окисляются и образуют смолы. Наличием смол и полимеров объясняется повышенная склонность крекинг-бензина к нагарообразованню в двигателях.
184
Образование смол замедляется введением в бензины специальных присадок — ингибиторов окисления.
Содержание серы в крекинг-бензинах из сернистых нефтей достигает 0,5—1,2% (масс), что в 5—8 раз превышает допустимое по стандарту на автомобильные бензины. Особенно опасны активные сернистые соединения — сероводород и меркаптаны. Очистка крекинг-бензинов от сероводорода проводится с помощью щелочной промывки.
Фракция, выкипающая при 200—350°С, называется газойлем термического крекинга. Она используется как компонент флотских мазутов, газотурбинного и печного топлива. После гидроочистки газойль может использоваться как компонент дизельного топлива.
Крекинг-остаток представляет собой фракцию, перегоняющуюся выше 350 °С. Он, как и прямогонный мазут, используется в качестве котельного топлива для теплоэлектростанций, морских судов, печей промышленных предприятий. Качество крекинг-остатка как котельного топлива выше, чем прямогонного сырья, так как крекинг-остаток имеет несколько более высокое теплосодержание, более низкую температуру застывания и вязкость. Особенно важно то, что у крекинг-остатков пониженная вязкость. Это облегчает условия транспортировки котельных топлив по системе подводящих трубопроводов и распыл их в форсунках.
Технологическая схема. Термическому крекингу подвергают различные виды сырья: от легкого прямогонного бензина до гудрона и тяжелых дистиллятов вторичного происхождения, получаемых при коксовании и каталитическом крекинге. Технологическая схема установки зависит от того, какое сырье на ней перерабатывается. Общим для всех установок термического крекинга является наличие трубчатой печи для подогрева сырья до необходимой температуры и сообщения тепла реакции. В целях обеспечения требуемой глубины превращения на большинстве установок, особенно при переработке тяжелого сырья, предусматриваются специальные реакционные аппараты, в которых сырье выдерживается определенное время при температуре реакции. На современных установках термического крекинга, как правило, применяется крекинг в рециркуляцией.
При крекинге дистиллятного сырья пределы перегонки фракции, возвращаемой на повторный крекинг, совпадают с пределами перегонки сырья. Но по углеводородному составу сырье и рециркулят заметно различаются. В рециркуляте меньше парафиновых углеводородов, а ароматических и нафтеновых больше. Рециркулят вследствие этого более термически устойчив.
При углублении крекинга тяжелого остаточного сырья на рециркуляцию возвращается фракция с пределами перегонки 350— 500 °С. В этом случае отгонять из продуктов крекинга все фракции, перегоняющиеся ниже температуры начала кипения исходного сырья, нельзя, так как после извлечения всех легких фракций крекинг-остаток становится очень вязким и непригодным для дальнейшего использования.
18S
Ниже представлена схема крекинга с рециркуляцией:
Свежее сырье
Крекинг
| Газ | ----- *- Газ | |||
| Бензин | ----- 9- Бензин | |||
|
|
| |||
| Крекинг- | ----- »■ Крекинг- |
| ||
| флегма ---- > KpcKitnr — | флегма | ------ >■ Крекинг и т. д, | ||
| Крекинг- | ----- У Крекинг- |
| ||
| остаток | оетяток | |||
Отношение количества рециркулята и свежего сырья называется коэффициентом рециркуляции К\- Отношение всей загрузки
ХК-1
Е-1 ХК-2
XK-J
л
= к-з Д,_ 1_
Е = ф.К-4
К-5
Ё-3
| Т-3 |
| к-\ |
| К-2- |
| &Е |
| Т-2 Lfg? |
У'г»
| I |
Н-З
| Q\ jQ |
mi
| IV |
| Ч^ |
h&u
| vv[ |
0-
| •3* |
Т-1,Х-4/,
| ш |
| й-1 Н-1 |
Н-1
| Н-2 |
-Н-З Н-4
Рис. 38. Технологическая схема установки термического крекинга:
/ — сырье; // — газ; ///—головка стабилизации; IV—стабильный бензин; V—кероеино-газойлевая фракция; VI— крекинг-остаток.
крекинг-печи к загрузке свежим сырьем носит название коэффициента загрузки Кг- Между /Ci и Ki существует зависимость
Чем больше глубина крекинга за однократный пропуск сырья, тем меньше коэффициент циркуляции и соответственно выше производительность установки по свежему сырью. Однако увеличение глубины крекинга за. однократный пропуск приводит к снижению выхода бензина, увеличению образования кокса и газа, повышению летучести легкой бензиновой фракции.
На рис. 38 приводится технологическая схема установки термического крекинга.
Тяжелый остаток (мазут или гудрон) насосом Н-1 забирают из промежуточных парков установки, затем он проходит через теплообменник Т-1, где подогревается за счет тепла крекинг-остатка, и затем делится на два потока. Один из потоков идет в нижнюю часть колонны К-3, а второй — в верхнюю часть испарителя
186
низкого давления К-4. Поток сырья, поступивший в К-4, обогащается там тяжелыми газойлевыми фракциями и насосом Н-2 подается в К-3. Сырье с низа К-3 печным насосом Н-9 подается в печь тяжелого сырья П-1.
Колонна К-3 разделена на две части «глухой» тарелкой. Через эту тарелку пары из нижней части переходят в верхнюю. Жидкость же из верхней части в нижнюю не поступает. Скапливающаяся на глухой тарелке жидкость забирается насосом Н-4 и подается на глубокий крекинг в печь легкого сырья П-2.
Предварительной подачей сырья в ректификационную колонну и смешением его с продуктами реакции одновременно решается несколько задач: используется тепло продуктов реакции, от сырья отгоняются легкие фракции, охлаждаются продукты реакции.
Продукты крекинга, выходящие из П-1 и П-2, объединяются н поступают в реакционную выносную камеру К-1, откуда переходят в испаритель высокого давления К-2. Здесь от парожидкостной смеси, которую, представляет поток, выходящий из печей, отделяется жидкость — крекинг-остаток. Крекинг-остаток самотеком поступает в испаритель низкого давления К-4.
В К-4 из крекинг-остатка за счет снижения давления выделяются пары газойлевой фракции. Подобно колонне К-3 испаритель К-4 делится на две части глухой тарелкой. Пары керосино-газойле-вой фракции, которые отделяются от крекинг-остатка в нижней' части К-4, через глухую тарелку попадают в верхнюю часть, где встречаются с движущимся навстречу жидким сырьем.. Контактируя с сырьем, пары керосино-газойлевой фракции частично конденсируются.
Несконденсировавшаяся часть газойля уходит с верха К-4 и, пройдя конденсатор-холодильник ХК-2, через емкость Е-2 выводится с установки. Часть газойля используется для орошения К-4.
Поток паров из верхней части испарителя К-2 поступает на разделение в ректификационную колонну К-3. С верха этой колонны уходят бензиновые фракции и газ. Верхний продукт К-3 охлаждается в конденсаторе холодильнике ХК-1 и разделяется.
С низа колонны К-3 уходят тяжелые фракции, которые совместно со свежим сырьем возвращаются на повторный крекинг. Эти фракции являются рециркулятом печи тяжелого сырья П-1.
Легкие газойлевые фракции, как уже указывалось выше, с глухой тарелки колонны К-3 поступают на крекинг в П-2.
На некоторых НПЗ установки термического крекинга реконструированы и работают в режиме легкого крекинга, предназначенного для получения маловязких котельных топлив — висбрекинга. Поскольку основная задача при висбрекинге состоит в том, чтобы до требуемого значения снизить вязкость, степень превращения сырья (чаще всего гудрона или полугудрона) ограничивают настолько, чтобы предотвратить образование карбенов и карбоидов и распад промежуточных фракций до бензина и газа.
Режим установки термического крекинга. Ниже приводится технологический режим установки термического крекинга:
187
Температура, "С Давление, МПа
Печь тяжелого сырья П-1
вход................................................. ...... 390—410 5,0—5,6
выход.............................................. ...... 470—490 2,2—2,7
Печь легкого сырья П-2
вход .......................................... ...... 290-320 5,5-6,5
выход.............................................. ...... 530-545 2,2—2,8
Реакционная камера К-1
веРх................................................... л^лт ) 0,85-1,2
низ................................................... 460—470 J
Испаритель высокого давления К-2
верх ........................................... 455—460 \ „ ак , „
низ................................................... 430-440 ) и.»5-1,2
Ректификационная колонна К-3
низ................................................... 390-410 -J
аккумулятор сырья........................ 280—320 > 0.8—1,2
верх ........................................... 210—220 J
Испаритель низкого давления К-4
низ................................................... 400—415 }
аккумулятор сырья........................ 280—300 > 0,15—0,3
верх ........................................... ...... 120—200 J
Для этой установки характерно высокое давление в аппаратуре. Высокое давление позволяет поддерживать более высокие температуры в колоннах и испарителях, что в свою очередь дает возможность повысить температуру поступления рециркулята в печь и уменьшить расход топлива на его подогрев до температуры крекинга. Благодаря повышенному давлению уменьшаются размеры аппаратов.
Чтобы затормозить протекание реакций крекинга, на выходе из трубчатой печи или выносного реакционного змеевика в линию парожидкостной смеси продуктов крекинга подается охлажденный продукт с температурой 160—165 °С.
Материальный баланс.Точный материальный баланс термического крекинга устанавливают в результате исследований на лабораторных и опытных установках.
Ниже приводится материальный баланс [в % (масс.)] установки термического крекинга при работе н проектном режиме на пря-могонном мазуте (фракция выше 350 °С) сернистой нефти (I) и смеси 75% гудрона (фракция выше 500°С) и 25% тяжелого газойля каталитического крекинга (II) и при работе в режиме вис-брекинга на гудроне (фракция выше 460°С) арланской нефти
(III):
i п ш
Поступило:
Сырье. ............................................................ 100,0 100,0 100,0
Получено:
Углеводородный газ......................................... 3,5 2,5 2,3
Головка стабилизации...................................... ........... 3,6 3,4 3,0
Крекинг-бензин................................................. ......... 19,7 14,2 6,7
Газойль (фракция 200-350 °С)........................ 5,3 3,9 -
Крекинг-остаток................................................ ......... 66,3 74,4 86,8
Потери............................................................... ........... 1.6 1,6 1,2
Итого ... 100,0 100,0 100,0
188
§ 39. ОСНОВНАЯ АППАРАТУРА
И ЭКСПЛУАТАЦИЯ УСТАНОВОК
ТЕРМИЧЕСКОГО КРЕКИНГА
Основные аппараты установки термического крекинга — трубчатые печи, реакционные камеры, испарители и ректификационные колонны.
Трубчатые печи установок термического крекинга предназначены не только для нагревания и частичного или полного испарения сырья, но и для проведения в них химических реакций. Этим они отличаются от печей перегонных установок.
В трубчатой печи крекинг-установки имеются зона нагрева и зона реакции. Установить точно границу между зонами невозможно, так как одновременно с разложением сырья происходит его дальнейшее нагревание. Участок труб, на котором завершается реакция крекинга, называется реакционным змеевиком.
Реакционный змеевик располагается в радиантной части печи, поскольку теплонапряженность труб в радиантной части печи в 2—3 раза больше, чем в конвекционной и, следовательно, можно добиться резкого уменьшения размеров змеевика.
Продукты крекинга, которые выходят из печи, несут много тепла, имеют высокую температуру. Это тепло используется для углубления реакций крекинга. Пары направляются в выносную реакционную камеру, которая представляет собой полый цилиндрический аппарат диаметром 2—3 м и высотой 10—15 м.
Вход паров в реакционную камеру предусмотрен сверху, а выход— снизу. Камера таким образом все время заполнена парами крекируемой смеси, и объем жидкости, в которой сконцентрированы наиболее легко коксующиеся продукты, невелик.
Так как реакционная камера не обогревается, а реакция крекинга идет с поглощением тепла, температура на выходе из камеры на 30—40 °С ниже, чем на входе. Вследствие довольно большого объема камеры продукт может долго (до 100 с) находиться в ней, а это способствует углублению крекинга. Как показал опыт эксплуатации, до 25% от общего количества бензина и газа образуется в реакционной камере.
Непосредственно из реакционной камеры парожидкостная смесь продуктов крекинга поступает в испаритель высокого давления. Так как разделение фаз в этом аппарате происходит при высоком давлении, в остатке, уходящем с низа испарителя, содержатся в большом количестве растворенные газой-левые и бензиновые фракции. Для выделения легких фракций предназначен испаритель низкого давления (фляшинг).
В результате снижения давления перед фляшингом происходит дополнительное испарение легких фракций из остатка. Чтобы улучшить условия испарения, в нижнюю часть фляшинга вводят водяной пар.
Испаритель высокого давления конструктивно представляет собой полый аппарат диаметром 2—3 м и высотой 15—18 м.
139
Испаритель низкого давления снабжен ректификационными тарелками. Он служит не только лля отго'нки легких компонентов из крекинг-остатка, но и для подогрева сырья за счет охлаждения и конденсации паров отпаренных фракций.
Технологический режим установки поддерживается в соответствии с технологическим регламентом и технологической картой.
Работу установки термического крекинга контролируют по показаниям приборов, на основании результатов анализа сырья, флегмы, крекинг-остатка и дистиллята. В сырье определяют содержание воды, сернокислотных смол, коксуемость, плотность. Коксуемость и содержание сернокислотных смол дают представление о способности сырья образовывать кокс в трубах печи и реакционной камере. Если на переработку поступает более смолистое сырье, то необходимо снизить температуру крекинга в пределах, установленных технологической картой.
Правильнее ведение технологического режима дает возможность увеличить продолжительность безостановочного пробега. При нормальных условиях эксплуатации безостановочный пробег крекинг-установки составляет 40—45 и даже 60 сут. Остановка установок термического крекинга вызывается необходимостью очистки их от кокса. Кокс, откладывающийся в трубах печи, уменьшает свободное сечение змеевика, что приводит к повышению давления на входе в печь. Заметные отложения кокса наблюдаются в нижней части реакционной камеры, испарителей, в трубопроводах крекинг-остатка. При интенсивном отложении кокса межостановочный пробег установки снижается до 25—28 сут.
Чтобы уменьшить отложения кокса, на установках термического крекинга внедрена подача в сырье перед печью небольшого количества турбулизирующего, т. е. ускоряющего скорость движения потока вещества — воды. Попадая в поток сырья, имеющий температуру 390—400°С, вода немедленно испаряется. Объем паровой фазы резко увеличивается, поток сырья интенсивно перемешивается, скорость потока возрастает. Особенно важно, что увеличивается скорость движения расположенной у стенки трубы граничной пленки. Именно эта пленка является основным источником коксообразования.
§ 40. НАЗНАЧЕНИЕ И ОСНОВНЫЕ ТИПЫ УСТАНОВОК КОКСОВАНИЯ НЕФТЯНЫХ ОСТАТКОВ
Назначение процесса коксования.Образование кокса при термическом крекинге ограничивает возможности дальнейшего углубления процесса. Выход светлых продуктов при крекинге гудрона или мазута не превышает 35—40%. Если же установка термического крекинга работает в режиме висбрекинга, то выход светлых продуктов еще меньше.
Выход их можно значительно повысить, если при термическом разложении не опасаться образования кокса, не считать его вредным побочным продуктом. Технической формой такого деструктивно
ного процесса является коксование — одна из разновидностей термических процессов.
При коксовании твердый углеродистый остаток — кокс является конечным продуктом разложения и образуется в значительных количествах. Наряду с коксом получают бензин, газоилевые фракции и газ.
Коксование позволяет утилизировать, превращая в светлые нефтепродукты, не только прямогонные остатки, но и такие продукты, как асфальты и экстракты масляного производства. То обстоятельство, что при этом получается в качестве товарного продукта кокс, является в настоящее время достоинством процесса.
Типы установок.Существуют установки для трех типов процессов коксования: периодические, полунепрерывные, непрерывные.
Периодический процесс — наиболее старый из существующих процессов коксования. Коксование проводится в обогреваемых кубах, которые представляют собой горизонтальные аппараты диаметром 2—4,5 м и длиной 10—13 м. Сырье загружают в куб и постепенно подогревают его сниау открытым огнем. При 300°С начинается выделение дистиллятных паров, которое усиливается по мере разогревания куба. После того как температура в паровой-зоне куба достигнет 445—460 °С, начинается ее снижение.
Понижение температуры говорит о том, что выделение погонов прекратилось и процесс образования коксового «пирога» в основном закончился. Дальнейший подогрев куба необходим для завершения процесса коксования, прокалки и подсушки кокса.
После прокалки, которая продолжается 2—3 ч, температуру в топке под Кубом постепенно снижают, выключают форсунку и охлаждают куб. Для охлаждения подается сначала водяной пар, а затем воздух. Когда температура кокса понизится до 150— 250 "С, приступают к его выгрузке. Выгрузка кокса из кубов почти не механизирована и представляет собой очень тяжелую, трудоемкую операцию.
По своему техническому уровню периодические кубовые установки давно устарели, однако некоторые коксовые кубы и батареи еще эксплуатируются. На кубовых установках получают кокс высокого качества, с низким содержанием летучих.
Полунепрерывный процесс коксования пришел на- смену кубовым установкам. Он проводится' в необогреваемых коксовых камерах (замедленное коксование). Этот процесс известен с начала 1930-х гг. В настоящее время замедленным коксованием получают наибольшее количество нефтяного кокса во многих странах мира.
Сырье замедленного коксования нагревается в трубчатой печи до 500 °С и направляется в полый необогреваемый вертикальный цилиндрический аппарат — коксовую камеру (реактор). В камере сырье находится длительное время и за счет аккумулированного им тепла коксуется. С верха работающей камеры удаляются потоки легких дистиллятов. После заполнения реактора коксом на
191
70—90% поток сырья переключается на другую камеру, а из отключенной камеры выгружается кокс.
Процесс замедленного коксования имеет периодический характер по выгрузке кокса и непрерывный — по подаче сырья и выделению дистиллятных продуктов.
При непрерывном коксовании нагретое сырье вступает в контакт с подвижным, нагретым до более высокой температуры инертным теплоносителем и коксуется на поверхности этого теплоносителя. Кокс, отложившийся на поверхности теплоносителя, вместе с ним выводится из зоны реакции. Затем теплоноситель и отложившийся на нем кокс поступают в регенератор, где часть кокса выжигается. За счет тепла, выделившегося при сгорании, происходит подогрев теплоносителя до -требуемой температуры. Нагретый теплоноситель возвращается в зону реакции.
Поскольку основное количество тепла, необходимого для коксования, сообщается за счет контакта сырья с нагретым теплоносителем, сырье перед подачей в реактор можно подогревать до более низкой температуры, чем требуется при замедленном коксовании. Это облегчает переработку наиболее высоковязких, смолистых продуктов, например асфальтов масляного производства, которые быстро коксуются при подогреве в трубчатых печах.
Ш работающих установках непрерывного коксования теплоносителем является порошкообразный кокс с размером частиц до 0,3 мм, а коксование происходит в кипящем слое теплоносителя. Порошкообразный кокс легко перемещается внутри установки (из реактора в регенератор и обратно), что позволяет строить установки большой мощности.
Для перемещения коксового теплоносителя используется принцип пневмотранспорта. Движущей силой является поток пара или газа, захватывающий коксовые частицы и несущий их. Существуют различные системы пневмотранспорта: в разреженном слое и в плотном слое.
При непрерывном коксовании в кипящем слое происходит одновременно три процесса: собственно коксование, сопровождающееся образованием продуктов разложения и уплотнения, прокалка кокса, благодаря которой из кокса удаляются летучие, и наконец вторичные реакции распада и уплотнения продуктов коксования, находящихся в паровой фазе.
Высокая температура коксового теплоносителя способствует испарению продуктов разложения и удалению их с поверхности коксовых частиц. Возможность образования продуктов вторичного происхождения уменьшается. Поэтому выход кокса при непрерывном коксовании меньше, чем при замедленном.
Выбор той или иной модификации процесса коксования для строительства на НПЗ зависит от того, какая цель при этом преследуется. Экономические показатели (себестоимость продукции, стоимость строительства, численность обслуживающего персонала) установок замедленного коксования и коксования в кипящем слое приблизительно одинаковы.
192
Достоинством замедленного коксования является больший выход кокса. При переработке, одного и того' же сырья замедленным коксованием можно получить в 1,5—1,6 раза больше кокса, чем при коксовании в кипящем слое. Освоенная технология прокалки кокса приспособлена только для переработки кускового кокса.
Установки замедленного, коксования строятся в тех случаях, когда необходимо удовлетворить потребность в нефтяном коксе, а установки коксования в кипящем слое — для увеличения выработки светлых нефтепродуктов переработкой остатков.
Сырье коксования.Коксованию подвергаются высокомолекулярные нефтяные остатки: гудроны, крекинг-остатки термического крекинга, асфальты и экстракты с установок масляного производства, смолы пиролиза. Основными показателями качества сырья являются коксуемость, содержание серы и золы, вязкость.
Остатки, являющиеся сырьем коксования, состоят из высоко
молекулярных углеводородов, смолисто-асфальтеновых веществ,
карбенов и карбоидов. Соотношение между составляющими сырья
зависит от происхождения нефти, температуры и продолжитель
ности процесса, при котором был получен остаток. Коксуемость и,
следовательно, выход кокса при коксовании тем выше, чем больше
в сырье смолисто-асфальтеновых веществ. Как, правило, коксуе
мость крекинг-остатка выше, чем гудрона. Экономически целесо
образно, чтобы коксуемость сырья была не менее 10%. Однако
если в сырье слишком много смолисто-асфальтеновых веществ и
коксуемость превышает 20%, то это приводит к быстрому за-
коксовыванию печей нагрева сырья, а значит и частым останов
кам установки. ''-
Для производства кокса, используемого в специальных целях, необходимо особенно тщательно подбирать сырье, используя для коксования крекинг-остатки, полученные термическим крекингом дистиллятных продуктов, смолы пиролиза и т. п.
Основные потребители нефтяного кокса нуждаются прежде всего в малосернистом коксе, поэтому на коксование следует.направлять остатки, содержащие не более 0,5—0,8% серы. В коксе содержится, как правило, в 1,5 раза больше серы, чем в сырье. Поскольку промышленно освоенных процессов очистки кокса от серы еще не существует, установки коксования сооружаются в настоящее время там, где имеется возможность обеспечить их малосернистым сырьем.
Состав и свойства продуктов.Газ по составу аналогичен газу термического крекинга, однако содержит несколько меньше оле-финовых углеводородов. При более жестком режиме коксования (повышенной температуре) содержание непредельных углеводородов в газе растет. Так, повышение температуры коксования в кипящем слое с 520 до 540 °G приводит к увеличению выхода непредельных углеводородов с 45 до 52%.
Бензин содержит много непредельных углеводородов, что делает его недостаточно химически стабильным. Октановое число бензинов замедленного коксования составляет 68—72 пункта.
7 Зак. 380
193
Керосино-газойлевые фракции (180—350 °С, 350—450 °С) используются как компоненты газотурбинного топлива и сырье каталитического крекинга. Фракция 180—350 °С может быть направлена в дизельное топливо.
Тяжелый газойль (фракция выше 450 °С) используется как компонент котельного топлива.
Нефтяной кокс широко применяется в различных отраслях народного хозяйства. Он используется для производства электродов, абразивных материалов, карбидов, углеграфитовых материалов, ферросплавов. Расход кокса при производстве различных продуктов составляет: алюминия — 0,6 т кокса на 1 т продукции; карбида кремния—'1,4 т/т; карбида кальция — 0,7 т/т; графита — 1,25 т/т. Чтобы удовлетворить непрерывно растущую потребность в нефтяном коксе, ежегодно строятся новые установки коксования. В настоящее время в мире вырабатывается свыше 15 млн. т/год нефтяного кокса. В нашей стране производство кокса за 10 лет (с 1966 г. по 1976 г.) увеличилось более, чем в 5,5 раза.
В зависимости от назначения к нефтяным коксам предъявляют различные требования. Основными показателями качества кокса
ТАБЛИЦА
Свойства коксов, полученных методом замедленного коксования из различных видов сырья
Показатели
Гудрон
котур-
тешшской
нефти
Крекинг-остаток волгоградских нефтей
Крекинг-остаток татарских нефтей
Гудрон
и асфальт
ферганских
нефтей
Сырье
Плотность р™...................
Коксуемость % .........
Содержание, %
серы................................
золы................................
Групповой состав, %
масла всего.....................
в том числе: парафины ......
смолы ...................
асфальтены.................
' карбены и карбоиды
Выход фракций, % (масс.)
выше 25 мм....................
ниже 25 мм....................
Содержание, 96 (масс.)
серы.................................
золы................................
летучих .....................
Механическая
прочность, МПа ....
0,958 9,2
0,47 0,08
68,6
5,9
5,9
29,9
Кокс
41,5 58,5
0,52 0,43—0,5 8,7—10,2
4,0-5,7
0,920 6,5
0.55 0,10
83,0
14,0 2,5 0,3
35,7 64,3
0,56 0,25-0,47 8,0-9,5
3,5-4,6
1,026 21,2
2,92 0,12
58,5
'26,9
14,4
0,2
45,0 55,0
4,0 0,46-0,49 6,8-7,2
5,3-6,0
0,993 14,3
0,62 0,10
67,0
25,5 7,5
45,0 55,0
0,92 0,38—0,42 6,8—9,3
9,0
194
являются содержание серы, золы, влаги, летучих, гранулометриче-' ский состав, реакционная способность, пористость, удельное электрическое сопротивление и др. По содержанию серы коксы делят на малосернистые (до 1,0% S), среднесерийные (до 1,5% S), сернистые (до 4,0% S), высокосернистые (выше 4,0% S); по гранулометрическому составу —на кусковой (фракция с размером кусков выше 25 мм), «орешек» (фракция 6—25 мм), мелочь (менее 6 мм); по содержанию золы — на малозольиые (до 0,5%. золы), средне-зольные (0,5—0,8%), высокозольные (более 0,8%).
Качество кокса, особенно по таким показателям, как содержание серы, летучих, золы, зависит от свойств сырья, что наглядно иллюстрируется данными табл 7. Выход и качество кокса зависят, также от процесса его получения. Характеристика коксов, полученных различными процессами коксования, приводится в табл. 8. Кубовый кокс лучше, чем кокс замедленного коксования, так как он содержит меньше летучих веществ, золы и влаги.
ТАБЛИЦА 8
Выход и характеристикакокса различных процессов коксования
Кокс
Выход
кокса
%(масс.)
Содержание, %(масс.)
Малосернистый
Кубовый
из крекинг-остатка . .
из пиролизного сырья .
Замедленного коксования
из гудрона
фракция выше 25 мм
(КЗ-25) ...................
6-25 мм (КЗ-6) . . . 0-6 мм (КЗ 0) . . . .
16-20 24-26
20-22
1-4 0,5-2,0
0,4-0,8 0,01-0,2
0,5-0,6 0,7 0,8
0,5-1,0 0,1-0,8
1,3-1,5 1,5 1,5
5-7 3-5
7-9 9,5 10
Сернистый
Кубовый из крекинг-остатка
Замедленного коксования
из гудрона
фракция выше 25 мм .
» ниже 25 мм .
Порошкообразный термо
контактного крекинга из
гудрона
24-30
22-24
10-12
1-4
6-8 6-9
0,5-2,0
0,4-0,8
0,3-0,4 0,3-0,5
0,6-0,9
3,3-5,3
3,4-3,8 3,5-3,9
3,4-4,0
5-7
5-7 9-10
2-3
Кокс, используемый в производстве электродов, подвергается дополнительной обработке — прокаливанию в специальных печах при 1200—1300°С. Прокалка может проводиться сразу же после получения кокса на НПЗ или у потребителя. При прокалке удаляются летучие, снижается электрическое сопротивление, устраняются усадочные явления. Прокалочные печи на алюминиевых
7*
195
и электродных заводах предназначены только для кокса с размером частиц выше 25 мм; коксовую мелочь, а она составляет около 50% кокса замедленного коксования, в этих печах прокаливать нельзя. Для прокалки коксовой мелочи сейчас используют камерные печи, ранее применявшиеся для получения искусственного газа из горючих сланцев на сланцеперерабатывающих предприятиях.
§ 41. ТЕХНОЛОГИЧЕСКИЕ СХЕМЫ И РЕЖИМ РАБОТЫ УСТАНОВОК КОКСОВАНИЯ
Замедленное коксование
Технологическая схема. Схема установки приводится на рис. 39. Сырье коксования подается насосом Н-1 через печи П-1 и П-2 в ректификационную колонну К-1 на верхнюю каскадную тарелку. Под нижнюю каскадную тарелку колонны К-1, конструкция которой аналогична колонне термического крекинга, подаются горячие пары продуктов коксования из реакционных камер. За счет контакта паров, имеющих температуру около 430 °С, с менее нагретым сырьем последнее подогревается. При этом часть паров конденсируется. Сконденсировавшиеся продукты коксования служат рецир-кулятом, вместе с первичным сырьем они уходят с низа К-1 в реакционные змеевики, расположенные в радиантной части трубчатых печей.
В печах сырье подогревается до температуры начала коксования (500—510 °С) и поступает через нижний загрузочный штуцер в реакционные камеры.
На установке имеются четыре камеры, работающие попарно, независимо друг от друга. Каждую пару камер можно отключать на ремонт, не останавливая установки. Сырье из П-2 подается в коксовую камеру Р-1 или Р-2, а из печи П-1 — в камеру Р-3 или Р-4.
Из камер продукты реакции направляются в ректификационную колонну К-1. Нижняя часть колонны снабжена каскадными тарелками, верхняя — ректификационными. В верхней части колонны происходит разделение продуктов реакции на фракции.
Бензиновые пары и газ уходят с верха колонны в конденсаторы-холодильники ХК-1. Конденсат из ХК-1 подается в водогазо-отделитель Е-1, где происходит отделение газа от бензина и бензина от воды. Вода сбрасывается в емкость Е-2 и затем используется для получения пара в специальном змеевике печи. Избыток воды переливается в канализацию. Бензиновая фракция и газ самостоятельными потоками направляются на дальнейшую переработку в газовый блок.
С 31-, 26-, 18-й тарелок К-1 отбираются боковые погоны: фракции выше 450 °С, 350—450 СС, 180—350 СС. В отпарной колонне К-2 из боковых погонов удаляются легкие фракции. Затем фракции с низа секций отпарной колонны через теплообменники и холодильники отводятся с установки. Часть фракции 180—350°С
W6
Ж-Г
CLHL
^л
| К |
чш
га
| Р-4 |
| X |
| а £-4 jit/ |
|
|
|
|
|
|
|
|
| Р-2 |
| к |
| П-2 |
Р-3
Н-2
Н-3 Н-4
3 н
Н-5
Я-; 7-г
T-JL
JT
Рис. 39. Технологическая схема установки
/ — сырье; // — сухой газ; /Л — головка стабилизации; IV— стабильн 350-450 °С; VII— фракция выше 450 °С; VII
используется как абсорбент в доабсорбере газового блока. Для снятия избыточной теплоты с 21-й тарелки К-1 выводится промежуточное циркулирующее орошение, которое после охлаждения возвращается в колонну.
Газовый блок состоит из абсорбера-десорбера, дополнительного абсорбера (доабсорбера), стабилизатора. Обработкой легких продуктов коксования с применением методов абсорбции и стабилизации (см. § 60) получают сухой газ, состоящий в основном из углеводородов Ci—С2, головку стабилизации, состоящую из углеводородов Сз—Q, и стабильный бензин. Стабильный бензин очищается от сернистых соединений щелочной промывкой и выводится с установки.
Реакционные камеры установки замедленного коксования работают по циклическому графику. В них последовательно чередуются циклы: реакции, охлаждения кокса, выгрузки кокса и разогрева камеры.
В начальный момент при подаче сырья в неработавшую камеру происходит разогрев ее стенок горячим сырьем. В этот период процессы испарения преобладают над крекингом, а дистиллят, уходящий с верха реакционной камеры, состоит из почти не подвергшихся разложению легких фракций сырья. В нижней части камеры накапливается жидкая масса, которая представляет собой тяжелую чаеть загруженного сырья.
Продолжительность первого периода зависит от качества сырья и от того, до какой температуры оно было подогрето. Так, для крекинг-остатков с высоким содержанием асфальтенов при температуре нагрева сырья 475 °С, первый период продолжается 5 ч, а при 510 °С — всего 2 ч; для полугудрона, содержащего меньше асфальтенов, продолжительность первого периода составляет при тех же температурах нагрева сырья соответственно 8—9 и 5—6 ч.
При дальнейшей работе подаваемое в камеру сырье проходит через все более высокий слой жидкости, в котором интенсивно происходят реакции деструкции. Вязкость жидкого остатка постепенно повышается, в нем накапливаются коксообразующие вещества и этот остаток постепенно превращается в кокс. Второй период коксования отличается постоянным выходом и качеством продуктов разложения.
Когда камера заполняется коксом примерно на 80%, поток сырья с помощью специального четырехходового крана переключают на другую камеру. В отключенной реакционной камере кок-сообразование из-за понижения температуры замедляется. В верхней части камеры в этот период откладывается губчатая рыхлая масса кокса. Всего по высоте камеры насчитывается три слоя кокса — нижний, образовавшийся в первый период, средний — во второй период и верхний слой, который откладывается в конце при охлаждении камеры. Кокс в верхнем слое наименее прочен, содержит много летучих соединений и обладает повышенной зольностью, так как он образовался за счет коксования смолисто-ас-
198
фальтеновых веществ. Зольные элементы и сера, как правило, концентрируются в этих веществах.
После отключения камеры, заполненной коксом, ее продувают водяным паром для удаления жидких продуктов и нефтяных паров. Удаляемые продукты поступают сначала в колонну К-1. После того как температура кокса понизится до 400—405 °С, поток паров отключается от К-1 и направляется в емкость Е-4. Водяным паром
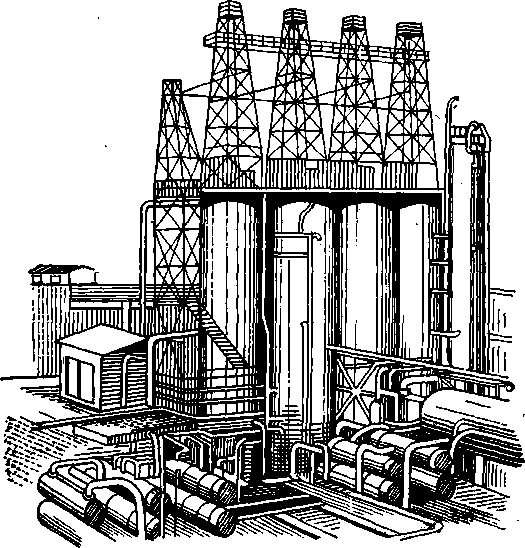
Рис. 40. Общий вид установки замедленного коксования.
кокс охлаждается до 200 °С, после чего в камеру начинают подавать воду. Вода подается до тех пор, пока вновь подаваемые порции воды не-перестанут испаряться. Сигналом о прекращении испарения служит появление воды в сливной трубе Е-4.
Закончив охлаждение, приступают к выгрузке кокса из камеры. Для очистки камер применяется гидравлический метод. Пласты кокса разрушаются струей воды, имеющей давление до 15 МПа. Над каждой камерой (рис. 40) установлены буровые вышки высотой 40 м, предназначенные для подвешивания бурового оборудования. Конструкция вышки и бурового оборудования, применяемого для удаления кокса, аналогична оборудованию для добычи нефти из скважин. На вышке закрепляется гидродолото, с помощью которого в слое кокса пробуривается центральное отверстие. Размеры отверстия достаточны для свободного прохода
199
гидрорезака. После пробуривания отверстия гидродолото снимают и заменяют гидрорезаком. Гидрорезак снабжен соплами, из которых подаются сильные струи воды, направляемые к стенкам камеры. Гидрорезак перемещается по камере, полностью удаляя со стенок кокс.
Дал^е'кокс транспортируется по схеме, приведенной на рис. 41. Удаленные из камеры кокс и вода через разгрузочный люк диаметром 1,4 м попадают в дробилку Д-1, где кокс дробится на куски размером не более 250 мм. Раздробленный кокс поступает
ЛЛЛЛ
N
Р-2
Р-3
Р-4
 у у у чк СК~ \
у у у чк СК~ \
СКА
5&шж^
Рис. 41. Схема транспортирования кокса на установке замедленного коксования. Р-1—.Р-4— реакционные камеры; Д-t — питатель-дробилка: СК-1 — СК-8 — скребковые конвейеры; С-1 — С-6— силосные башни склада кокса; П-1-~П-6— питатели; Ф-I — фильтр-отстойник ;
В —железнодорожный вагон.
на скребковый конвейер СК-1, снабженный обезвоживающими днищами. Через днища вода удаляется, а кокс элеватором подается в двухоитовой грохот, где сортируется на три фракции: выше 25 мм, 25—6 мм, 6—0 мм.
Вода, удаленная через обезвоживающие днища, содержит коксовую мелочь, которую в специальных фильтрах-отстойниках отделяют от воды и переносят грейферным краном в бункер коксовой мелочи. Вода из отстойников возвращается в цикл.
Коксовую камеру, из которой выгружен кокс, опрессовывают и прогревают острым водяным паром. Подаваемый в камеру пар вытесняет находящийся в ней воздух. Затем в течение некоторого времени через камеру пропускают горячие пары продуктов коксования из работающей камеры. Пройдя через подготавливаемую камеру, эти пары поступают затем в ректификационную колонну К-1. Когда подготавливаемая камера прогреется до 360 °С, заканчивается образование кокса в работающей камере. В этот момент камеры переключают.
200
Технологический режим процесса
Температура, °С
первичного сырья на выходе из печи ....... 375—390
вторичного сырья на выходе из реакционного змее
вика ......................................................................... 495—506
паров на выходе из камеры............................................. .... 440—455
Давление в коксовой камере, МПа .... .............. 0,18—0,4
Коэффициент рециркуляции................................................... 1,2—1,6
Примерный график работы коксовой камеры (в ч)
Подача сырья . .................................................................... .......... 24
Переключение камеры............................................................ .......... 0ф
Пропаривание камеры водяным паром................................. 2,5
Охлаждение кокса водой......................................................... 3
Слив воды................................................................................ 2,5
Бурение отверстия в коксе гидродолотом............................ .......... 0,6
Расширение отверстия для прохода гидрорезака .... 1,6
Удаление кокса гидрорезаком................................................ .......... 4
Испытание камеры.................................................................. 2
Разогрев камеры...................................................................... .......... 7,5
Итого ... 48
Материальный баланс.Ниже приводится материальный баланс установки замедленного коксования при работе на гудроне жирновской (I) и ромашкинской (II) нефти и крекинг-остатке ромашкинской нефти (III).Наибольший выход кокса наблюдается при коксовании крекинг-остатка, который" содержит больше смо-листо-асфальтеновых веществ, чем гудрон.
i и ш
Характеристика сырья
Плотность р".......................................... 0,956 1,000 1,080
Коксуемость,, % (масс.) . ...... 8,2 17 23
Содержание серы, % (масс.) .... 0,68 3,1 3,4
Выход продуктов, %
Углеводородный газ ......... 4,2 5,4 5$
Головка стабилизации........................... ......... 2,4 3,2 1,9
Бензин..................................................... ........ 10,2 13,0 5,5
Легкий газойль....................................... 43,5 27,0 24,3
Тяжелый газойль................................... ........ 24,2 24,4 27,0
Кокс........................................................ 12,5 24,0 33,0
Потери..................................................... 3,(1 3,0 3,0
Итого .... 100,0 100,0 J 00,0
Аппаратура.Основным реакционным аппаратом на установке замедленного коксования является реакционная (коксовая) камера, представляющая собой пустотелый аппарат диаметром 3,0—7,0 м и высотой 22—30 м. Корпус и днища камер изготавливаются из биметалла (углеродистая сталь -f- сталь с содержанием 11—13%хрома).
Число и размер камер зависят от производительности установок по сырью, еп^ качества и давления в камерах. Повышение давления способствует увеличению выхода кокса и газа, позво-
201
ляет повысить скорость подачи жидкого сырья в камеру, использовать избыточное давление в системе для подачи газа на газовый блок.
Ректификационные колонны, как и на установках термического крекинга, не имеют отгонной части. Нижняя часть колонны представляет собой конденсатор смешения, в котором происходит теплообмен и массообмен между нагретым сырьем, поступающим из печей, и парами продукта из коксовых камер. Такое решение позволяет утилизировать теплоту продуктов реакции, поскольку на горячих потоках, идущих из камер, нельзя ставить теплообменники.
Техника безопасности.Наиболее серьезные неполадки на установках замедленного коксования возникают при нарушении режима в камерах и колонне К-1. К такого рода неполадкам относятся: 1) перебросы через верх реактора, которые происходят при накоплении в камере жидких продуктов, не подвергающихся при данной температуре разложению и коксованию, и образующих вспененную массу; 2) забивание нижней части колонны коксовой мелочью; 3) самовоспламенение кокса и «хлопки» в реакционной камере, которые имеют место, если после выключения реакционной камеры на разгрузку на длительное время были оставлены открытыми люки, а к разгрузке не приступали; 4) задержка жидкого остатка в камере и застывание его; 5) нарушения в работе печного насоса, приводящие к закоксовыванию реакционного змеевика в печи.
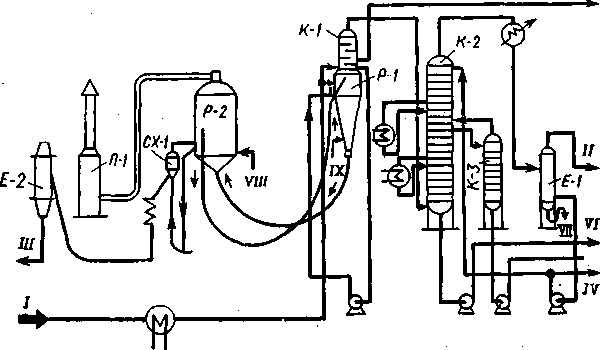
Коксование в кипящем слое
Процесс коксования в кипящем слое, иначе называемый термо-контактным крекингом (ТКК), в нашей стране освоен в опытном масштабе.
Технологическая схема.Схема установки ТКК приводится на рис. 42. Нагретое в теплообменниках сырье подается в парциальный конденсатор К-1, расположенный над реактором и представляющий собой одно целое с ним. В К-1 за счет тепла продуктов коксования, поступающих из реактора, от мазута, служащего сырьем, отгоняется широкая фракция, соответствующая по пределам перегонки (350—500 °С) вакуумному газойлю установок АВТ. Эта фракция вместе с парами продуктов коксования уходит на разделение в колонну К-2. Фра'кцию, перегоняющуюся выше 500 °С, забирают насосом с низа К-1 и подают на коксование в реактор Р-1.
Продукты коксования из Р-1 переходят в парциальный конденсатор К-1, где отделяется наиболее тяжелая часть продуктов реакции, которая идет на повторное коксование или выводится с установки как компонент котельного топлива.
В основной ректификационной колонне установки К-2, куда поступают пары из К-1, происходит разделение продуктов коксования на тяжелый газойль, легкий газойль и смесь бензина и газа.
202
В составе установки предусмотрены4 также блок очистки газа от сероводорода, сжатие газа компрессором и блок абсорбции и стабилизации, где проводится стабилизация бензина, выделение из газа головки стабилизации и деэтанизация последней.
Кокс-теплоноситель установки коксования в кипящем слое представляет собой порошок, состоящий из частац округлой формы диаметром 0,075—0,3 мм. Вместе с отложившимся на нем свежим коксом теплоноситель выводится снизу реактора и транспортируется в коксонагреватель Р-2, где также поддерживается
VI
|
|
| •-V |
Рис. 42. Схема установки термоконтактного крекинга (коксования в кипящем слое):
/ — сырье; II — газ на очистку от сероводорода и компримирование; III — кокс; IV—бензин в блок абсбрбции и стабилизации; V —легкий газойль (фракция 180—350 °С); VI — тяжелый газойль (фракция выше 350 °С); VII — вода; VIII — воздух; IX — водяной
пар.
взвешенный слой теплоносителя, но уже с помощью воздуха. Часть кокса в токе воздуха сгорает, а остальная нагревается до 600— 62dcC. Нагретый кокс возвращается в реактор.
Количество сжигаемого кокса Колеблется в зависимости от варианта работы установки и температуры реакции. Как показал расчет теплового баланса установки, сжигается около 40% от общего количества 'получаемого кокса.
При сжигании кокса образуются дымовые газы, содержащие значительное, количество окиси углерода и имеющие высокую температуру. В котле-утилизаторе П-1 этот газ сжигается и вырабатывается водяной пар. Количество пара превышает необходимое для нужд установки термоконтактного крекинга, и, следовательно эта установка служит как бы дополнительной котельной для НПЗ.
Часть кокса из Р-2 выводится в сепаратор-холодильник СХ-1, где крупные частицы кокса отделяются, охлаждаются и выводятся в качестве балансового кокса через емкость Е-2,
203
§ 42. СЫРЬЕ, ПАРАМЕТРЫ И ПРОДУКТЫ ПРОЦЕССА ПИРОЛИЗА
Назначение процесса.Пиролиз — наиболее жесткий из термических процессов переработки нефти. Он проводится пр.и температурах 750—900 °С и предназначается в основном для получения высокоценных олефиновых углеводородов — сырья нефтехимического синтеза.
Сырье.Наилучшим видом сырья для получения олефинов в процессе пиролиза являются парафиновые углеводороды. При распаде нормальных парафинов имеют место следующие основные закономерности: этан почти полностью превращается в этилен, из пропана и бутана с большим выходом образуются этилен и пропилен, из парафиновых углеводородов с числом углеродных атомов более 4 получают 45—50% (масс.) этилена, пропилен и непредельные углеводороды С4 и выше. При пиролизе- изопарафинов выход этилена меньше, образуется больше газообразных парафиновых углеводородов и в особенности метана. Ароматические углеводороды при умеренных температурах пиролиза являются балластом, а при более жестких в значительной степени образуют кокс и смолу.
Пиролизу могут подвергаться природные и попутные газы, а также продукты переработки нефти.
Природные газы содержат 93—98% метана, до 2% этана и до 1% пропана. Поскольку для пиролиза пригодны только этан и пропан, природные газы считаются малоценным сырьем пиролиза. Попутный газ и продукты стабилизации нефти содержат 60—70% этана, пропана и более высококипящих парафиновых углеводородов и поэтому представляют собой ценное пиролизное сырье. Иногда пиролизу подвергают не весь попутный газ, а только выделенные из него этан, пропан и бутан.
К продуктам нефтепереработки, применяемым для пиролиза, относятся прямогонный бензин и бензин-рафинат с установок экстракции ароматических углеводородов (см. § 54). При пиролизе бензиновых фракций дополнительно получают высокоценные продукты—бутадиен, бутилены, ароматические углеводороды, сырье для производства сажи и нафталина. Прямогонные бензины обладают преимуществами в сравнении с рафинатами, так как содержат в основном нормальные парафиновые углеводороды, тогда как в рафинатах — до 50% изопарафинов.
В перспективе возможно использование в качестве сырья пиролиза в трубчатых печах керосиио-газойлевых фракций. Однако это сырье нуждается в предварительной подготовке — очистке от сернистых соединений и деароматизацин.
В Советском Союзе на установках пиролиза перерабатываются все возможные виды сырья. Новые крупнотоннажные этиленовые установки ориентированы на переработку бензинового сырья.
Параметры процесса. Технологические параметры процесса-зависят от перерабатываемого сырья и того, какие углеводороды
204
необходимо получитьпри пиролизе. Оптимальнымсчитается режим, при котором обеспечивается максимальный выход олефинов. Важными параметрами процесса являются температура пиролиза, время контакта, парциальное давление паров сырья. Так, при пиролизе пропана с повышением температуры растет выход этилена и пропилена (рис. 43). Поскольку выход пропилена достигает максимума при более низкой температуре, изменением этого
| 'а? |
| 5! ? 20 |
| 1 |
| <з ю- |
| 1000 |
| зо- |
40 г
| 1у | „ / У\ | ||
| 2к/ у > / У | |||
| / У / У / У | |||
| - | (У | ||
| - у | l^N\J | ||
| &г | |||
| ^ | i | I I I | L.- 1 |
700 800 900
Температура ,°С
"0,1 0,7 0,4 0,8 2 4 в 20 Время контакта,с
Рис. 43. Зависимость содержания
олефинов (—) и водорода (---- )
в газе пиролиза пропана от температуры: / — всего олефинов; 2—этилен; Я—про-
Рис. 44. Зависимость выхода продуктов шролиза пропана от времени контакта:
/ — метан; 2— этилен; 3 — водород; 4 — пропилен; 5 —этан; 6 — углеводороды С4; 7—углеводороды С5; 8 — ароматические углеводороды.
параметра можно регулировать соотношение и выход этилена и пропилена.
Соотношение этилена и пропилена можно также регулировать, изменяя время контакта (рис. 44). Выход этилена повышается и при понижении парциального давления сырья. Процесс пиролиза проводится при давлении, близком к атмосферному, а парциальное давление регулируют, разбавляя сырье водяным паром. Разбавление сырья водяным паром уменьшает вероятность столкновения между собой молекул олефинов, и в результате снижается роль реакций полимеризации и уплотнения. При пиролизе газообразного сырья и сжиженных газов к сырью добавляют 10—20% водяного пара, при пиролизе бензинов и более тяжелых углеводородов— от 25 до 300%, считая на сырье.
Состав и свойства продуктов.При пиролизе образуются пиро-лизный газ и жидкие продукты пиролиза (смола пиролиза').
Пиролизный газ содержит большое количество различных компонентов. Его состав зависит от температуры пиролиза, времени пребывания в реакционной зоне (времени контакта) и качества исходного сырья. При большом времени контакта (больше
205
^v
2—3 с) характер сырья не оказывает заметного влияния на состав пиролизного газа. Режим процесса подбирается в зависимости от того, какой продукт пиролиза является целевым. При пиролизе газообразных углеводородов температуру процесса необходимо поддерживать более высокой, чем при пиролизе бензинового сырья.
Пиролизный газ на блоках газоразделения установок пиролиза делится на водород, метан, этан, этилен, пропилен, пропащ, бутилен-бутадиеновую фракцию. Из бутилен-бутадиеновой фракции выделяют бутадиен-1,3 — сырье промышленности синтетического каучука.
Смола пиролиза. Смолой пиролиза называют получаемые в процессе жидкие углеводороды от С5 и выше. Количество пиролиз-ной смолы в основном зависит От сырья пиролиза, что видно из следующих данных:
Выход смолы, % (масс) на сырье
Этан............................................................. 2—3
Пропан........................................................ ............. 8—10
Бутан............................................................ 5—8
Легкий бензин (п. к. —145 °С) .... 20—25
Керосино-газойлевая фракция .... 30—35
Выход смолы увеличивается также при снижении температуры пиролиза. Так, при низкотемпературном (740 °С) пиролизе выход смолы составляет 30—45%, а при высокотемпературном (800 °С) снижается до 5—20%.
Смола пиролиза содержит 10—15% диеновых углеводородов, 10—15% олефинов, 25—30% бензола, 10—15% толуола, а также непредельные соединения типа стирола и индена и циклоолефи-ны — циклопентадиен и др. Переработка смолы пиролиза может осуществляться по двум вариантам — топливному и химическому.
При топливном варианте смола дел:ится на две фракции — легкую и тяжелую. Из легкой фракции гидрированием удаляются непредельные углеводороды, очищенный продукт, называемый гидростабилизированным бензином, имеет октановое число 74— 78 пунктов и используется как компонент товарного автобензина. Тяжелая фракция направляется в котельное топливо.
При химическом варианте смола делится на фракции н. к. — 70 °С, 70—130 °С, 130—160 °С, 160—200 °С, 200—230 °С, выше 230 °С. Из фракции н. к.—70 °С выделяют циклопентадиен и изопрен, из фракции 70—130 °С — бензол, толуол и ксилолы. Разработаны методы гидрогенизацион'ной переработки этой фракции, позволяющие получать только бензол — наиболее ценный из ароматических углеводородов. Фракции 130—160 °С и 160—200 °С подвергаются полимеризации с получением синтетических нефте-полимерных смол — стирольной и кумарон-инденовой. Фракция 200—230 °С может быть использована как сырье для получения нафталина, а фракция выше 230 °С — для получения технического углерода (сажи).
206
§ 43. УСТАНОВКА ПИРОЛИЗА
Современная установка пиролиза состоит из следующих блоков: пиролиза углеводородного сырья, компрессии и очистки пиро-лизного газа, разделения газа, переработки смолы пиролиза.
Технологическая схема.Схема установки пиролиза бензиновых фракций приводится на рис. 45. Сырье под давлением 1,0—1,2 МПа подается в паровой подогреватель Т-1, нагревается до 100°С и после смешения с перегретым водяным паром поступает в конвекционную камеру печи пиролиза П-1. Здесь бензин испаряется и ,нагревается до 500—600 °С, после чего поступает в радиантную камеру, где протекает разложение углеводородов.
Выходящий из печи с температурой 750 °С и давлением 0,2— 0,22 МПа пиролизный газ направляется в закалочную камеру А-1, в которой быстро охлаждается до 700 °С за счет испарения подаваемого в эту же камеру парового конденсата. Быстрым охлаждением предотвращаются вторичные реакции полимеризации и конденсации олефинов.
Затем газы пиролиза охлаждаются до 400 °С в закалочно-ис-парительном агрегате Т-2, где их тепло используется для выработки водяного пара. Из Т-2 газы проходят в аппарат Е-1, куда впрыскивается поглотительное масло с температурой 70 °С. Газы пиролиза охлаждаются до 180 °С и направляются на дальнейшую переработку. В составе установки имеется несколько печдых агрегатов, состоящих из теплообменника, печи пиролиза, закалочной камеры, закалочно-испарительного агрегата и аппарата для впрыска масла. Охлажденные пиролизные газы от всех печных агрегатов объединяются в общий коллектор и направляются на промывку в колонны первичной ректификации К-1- В нижней части К-1 газы отмываются от сажи и кокса тяжелым поглотительным маслом, а в верхней — охлаждаются за счет испарения подаваемого туда легкого поглотительного масла. При этом происходит конденсация тяжелой смолы.
Из К-1 газы пиролиза поступают в водяные конденсаторы-холодильники ХК-1, где происходит конденсация легкого масла и водяных паров. В сепараторе С-1 конденсат легкого масла и вода отделяются от пиролизных газов, поступающих затем в блоки компрессии и очистки газа и газоразделения.
Тяжелое поглотительное масло с низа К-1 перекачивается насосом Н-1 в фильтры Ф-1, предназначенные для очистки от сажи и кокса. Очищенное масло делится на два потока, один из которых направляется на орошение нижней части К-1, а второй — охлаждается до 70 °С и подается в Е-1. Часть тяжелого поглотительного масла, в котором содержится также уловленная в К-1 тяжелая смола пиролиза, выводится на склад или в отделение переработки смолы.
Легкое масло и конденсат водяного пара из С-1 поступают самотеком в С-2, где происходит отделение масла от конденсата. Легкое масло, содержащее также легкую смолу пиролиза, делите
а
|
|
|
|
|
|
|
|
|
|
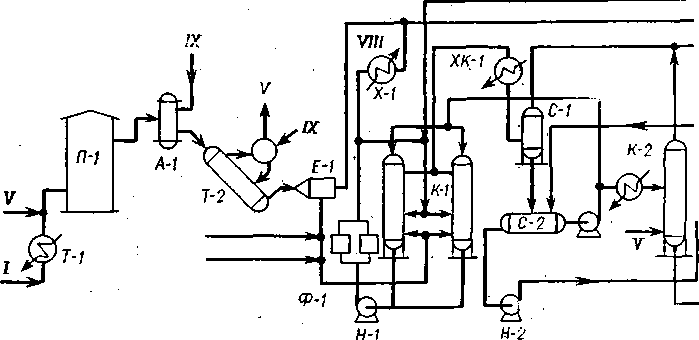
Рис. 45. Технологическая схема блока пиролиза углев /—сырье (бензиновая фракция); // —пиролизный газ на блок компресси К —водяной пар; VI — свежее поглотительное масло; VII — вода на о
питательная вода.
Mi
на два потока. Один из потоков возвращается на орошение верхней части К-1, а другой — подается в отпарную колонну К-2. В К-2 из масла отпаркой водяным паром удаляются легкие углеводороды — С4 и ниже, которые затем добавляются к газам пиролиза. Легкое масло с. низа К-2 насосом Н-3 выводится на склад или в блок переработки смолы пиролиза.
Конденсат водяного пара из С-2 загрязнен углеводородами, для очистки от них он подается на фильтры Ф-2. После фильтрации часть конденсата посылается на охлаждение пиролизных газов в закалочную камеру, • а избыток сбрасывается в канализацию. Отделенные на фильтре Ф-2 легкие углеводороды, содержащие некоторое количество водяного пара, конденсируются в ХК-2 и разделяются на водную и углеводородную фазы в сепараторе С-5.
Технологический режим процесса. Оптимальные режимы пиролиза различных видов сырья приводятся в табл. 9 (см. стр. 210).
Материальный баланс пиролиза прямогонного бензина (I) и этановой фракции (II) [в %(масс.)]
i и
Поступило:
Сырье............................................................................ 100,0 100,0
Получено:
Метано-водородная фракция...................................... 15,8 14,0
Этилен :....'............................................................... 24,5 50,0
Пропилен........................................................... . . . 15,0 1,0
Этам-пропановая фракция........................................... 7,5 31,0
Бутилен-бутадиеновая фракция.................................. 12,2 2,0
Жидкие продукты (С5 и выше).................................... ............ 25,0 2,0
легкая смола.......................................................... ........ (22,2) —
тяжелая смола....................................................... .......... (2,8) —
Итого ... 100,0 100,0
Аппаратура.Трубчатый пиролизный реактор (печь пиролиза)—основной реакционный аппарат современных пиролизных установок. Главные положительные качества трубчатого реактора — простота конструкции, небольшие эксплуатационные расходы, устойчивость работы.
Количество печей пиролиза на установке зависит от единичной мощности печи и производительности установки. Так, на установке пиролиза мощностью 250 тыс. т/год по сырью имеется шесть печей, в каждой из которых пиролизуется 7—7,5 т/ч сырья. В то же время на укрупненной пиролизной установке мощностью 1 млн. т/год сырья имеется всего четыре трубчатых печи, однако большей производительности.
В пиролизной печи должен быть создан такой объем реакционной зоны, чтобы сырье разлагалось до необходимой глубины в минимально короткий отрезок времени. Реакционный змеевик расположен в радиантной камере, где, как известно, теплонапря-женность поверхности нагрева максимальная.
209
ТАБЛИЦА 9
Оптимальные условия пиролиза различного сырья в трубчатых печах
| Температура, | Время | Концентрация | ||
| Сырье | Режим | °С | контакта, с | водяного пара, % |
| Этан | Этиленовый * | 825-830 | 1,0 | 10 |
| Пропан | » | 800 | 1,0-1,5 | 15 |
| Пропиленовый | 776 | 1,0 | 15 | |
| Бутан | Этиленовый | 800 | 0,5-1,0 | 20 |
| Пропилен-бутилено-вый | 750 | 1,0 | 20 | |
| Газовый бензин | Этиленовый | 780-800 | 1,0 | 20-50 |
| ^ | Пропиленовый | 750 | 1,0 | 20—50 |
| Прямогонный бен- | Этиленовый | 780-800 | 0,5-1,0 | 30-50 |
| зин | Пропиленовый | 750 | 0,5 | 25 |
| Бутиленовый | 725 | 1,0 | 25 |
* Этиленовым называется режим ведения процесса пиролиза, при котором получаются максимальные выходы этилена. Соответственно пропиленовый считается такой режим, при котором добиваются максимального выхода пропилена.
На пиролизных установках применяются трубчатые печи различной конструкции:
1) однокамерные, с конвекционной секцией, расположенной под подом печи, и горелками факельного типа;
2) двухкамерные, имеющие две топочные камеры, каждая из которых раздельно обогревается короткофакельными горелками; у этих печей общие для двух камер конвекционная камера и боров для вывода дымовых газов;
3) трубчатые печи с горелками беспламенного типа; существуют печи с экранами одностороннего и двустороннего облучения.
Конструкция печи с экранами двустороннего облучения и горелками беспламенного типа приведена на рис. 46. Производительность печи 6—8 т/ч сырья.
Теплота в радиантной части этой печи передается змеевику от излучающих стен топки. Стены собраны из беспламенных горелок конструкции ВНИИнефтемаша. Каждая горелка состоит из 20—25 керамических огнеупорных блоков с 4—6 туннелями в блоке. Газ, используемый для отопления, смешивается с воздухом и подается в туннели, длина которых составляет 10—15 см. Туннель имеет форму конуса, газ при движении по нему завихряется и быстро сгорает. При сгорании стенки туннеля раскаляются и излучают тепло в топку печи. Интенсивность теплоотдачи от раскаленных стен в 2,—2,5 раза выше, чем от факела, создаваемого обычной горелкой.
Температура газов, покидающих топку с беспламенными горелками, на 150—200 "С выше, чем в печах с факельными горелками.
210
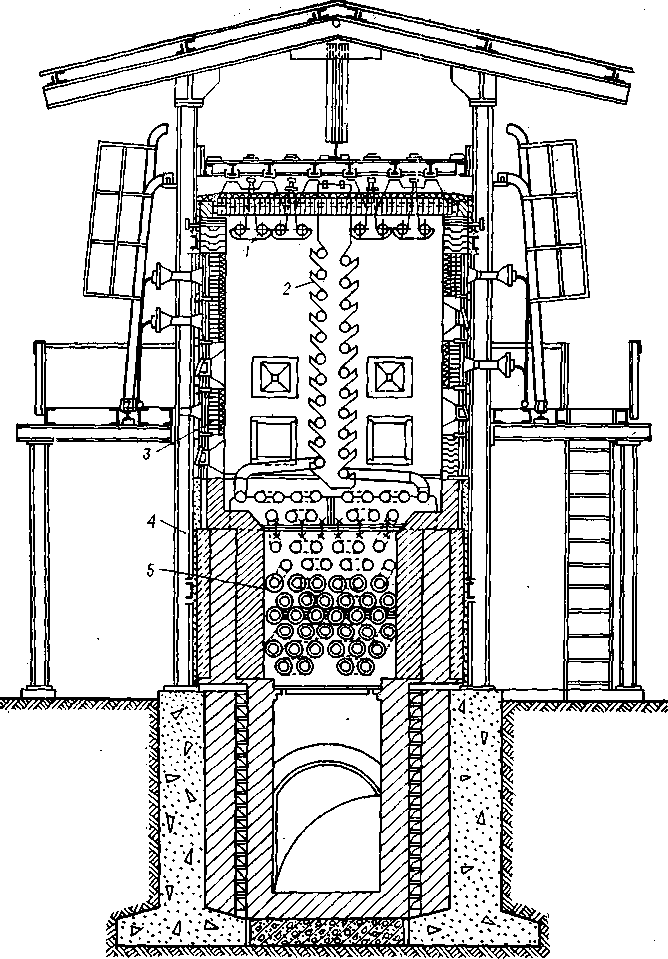
Рис. 46. Трубчатая печь пиролиза с экранами двустороннего облучения: / — потолочный экран; 2—экран двустороннего облучения; 3 — панельные горелки; 4 — каркас;
5 —конвекционная камера.
В печи с экранами двустороннего облучения трубы радиант-ной секции получают тепло с двух сторон и устраняется неравномерность обогрева змеевика. Тепловое напряжение в этих печах заметно выше, чем в печах одностороннего облучения: 40 кВт/м2 против 30—32 кВт/м2.
|
|
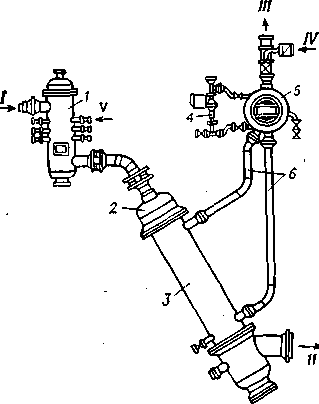
Закалочно-испар и тельный агрегат (рис. 47) представляет собой трубчатый теплообменник, по трубному пространству которого проходит пиролиз-ный газ, а в межтрубное подается промежуточный теплоноситель или специально подготовленная химически очищенная вода. Межтрубное пространство соединено трубопроводами с барабаном-паросборником.
| Рис. 47. Закалочно-исиарительный агрегат: / — газы пиролиза; // — охлажденные газы; /// — водяной пар высокого давления; IV — питательная вода; V —продувочная линия. 1 — закалочная камера; 2—передняя камера испарительной части; 3—теплообменник-испаритель; 4—выходная камера; 5 — барабан-паросборник; 6—трубопроводы для циркуляции воды. |
Автоматизация процесса. Установки пиролиза оснащены приборами и системами автоматического регулирования процесса. Давление паров в испарительной секции поддерживается автоматически подачей в теплообменник-испаритель греющего водяного пара с помощью регулятора давления. Температура газов пиролиза на выходе из пиролизных змеевиков регулируется изменением подачи топлива в печь. Очень важно своевременно изменить температуру пиролиза при изменении нагрузки печи и состава сырья. В настоящее время внедряются схемы регулирования с применением хроматографов. На основании хроматографическо-го анализа состава сырья автоматически изменяется режим. Автоматически регулируется также подача воды на закалку в зависимости от температуры пиролизного газа.
Перспективы дальнейшего развития процесса пиролиза. Пиролиз легких углеводородных фракций и средних (керосино-газой-левых) дистиллятов в трубчатых печах является в настоящее время наиболее надежным и экономически выгодным способом производства низших олефиновых углеводородов. Основные тенденции в развитии технологии процесса трубчатого пиролиза следующие:
1) переход к . строительству установок большой единичной мощности — до 1 млн. т/год сырья и более;
2) применение новой высокопроизводительной аппаратуры — печей с высокой тепловой напряженностью и зонным регулированием температуры по длине змеевика;
212
3) повышение селективности процесса за счет увеличения температуры процесса до 840—870 °С и снижения времени контакта до 0,2-0,4 с;
4) комплексное использование всех продуктов пиролиза — выделение из смолы пиролиза ароматических углеводородов, дивинила, изопрена, циклопентадиена с целью их дальнейшего использования;
5) расширение сырьевой базы — широкое применение бензи-
нов-рафинатов, переход к пиролизу керосино-газойлевых фрак
ций.
Методу получения этилена и пропилена пиролизом в трубчатых печах присущ наряду с несомненными достоинствами ряд недостатков. Для конструирования печей приходится применять большое количество высоколегированных сталей, температуру пиролиза, опасаясь разрушения труб печи, не поднимают выше 900 °С, на установках трубчатого пиролиза нельзя перерабатывать тяжелые виды сырья, чтобы не вызвать быстрого закоксовы-вания труб.
Поэтому внимание науки, проектных и промышленных организаций во всем мире привлечено к созданию новых методов пиролиза. Особый интерес вызывают такие модификации, которые позволяют перерабатывать тяжелые дистилляты, остаточные продукты и сырую нефть.
Новые процессы пиролиза тяжелых компонентов и нефти различаются по способу подвода тепла в реактор. Существуют, такие типы новых процессов пиролиза: 1) термоконтактные процессы; 2) пиролиз с перегретым водяным паром; 3) окислительный пиролиз; 4) пиролиз в плазме.
В свою очередь процессы термоконтактного пиролиза делятся на две группы. К первой относятся те, в которых применяется крупногранулированный теплоноситель. Вторую группу составляют процессы с использованием порошкообразного теплоносителя.
Общей чертой всех процессов термоконтактного пиролиза является наличие теплоносителя, который нагревается в специальном аппарате. Затем нагретый теплоноситель транспортируется в реактор, где вступает в контакт с пиролизным сырьем. К теплоносителю предъявляются требования высокой теплоемкости, механической и термической прочности, низкой абразивной способности. В качестве теплоносителя применяются шамот, кокс, кварцевый песок и др.
Новые процессы пиролиза в настоящее время проходят опытно-промышленную проверку,
Глава VII
ТЕРМОКАТАЛИТИЧЕСКИЕ ПРОЦЕССЫ ПЕРЕРАБОТКИ НЕФТЯНЫХ ФРАКЦИЙ
§ 44. ОСНОВНЫЕ ПРЕДСТАВЛЕНИЯ О КАТАЛИЗЕ И СВОЙСТВАХ КАТАЛИЗАТОРОВ
Большинство химических превращений углеводородов нефти, имеющих практическое значение, осуществляется в присутствии катализаторов. Катализаторы позволяют снижать энергию активации химических реакций и тем самым значительно повышать их скорость. В самом общем виде в этом и заключается сущность и значение катализа. Проведение реакции в присутствии катализаторов позволяет также резко снижать температуру процесса. Для реакций, характеризующихся положительным тепловым эффектом (полимеризация, гидрирование, алкилирование и др.), это имеет особо важное значение, так как высокие температуры с термодинамической точки зрения для них неблагоприятны. Следовательно, катализаторы в данном случае и ускоряют процесс, и способствуют достижению наиболее высоких равновесных концентраций. Следует, однако, не забывать, что сдвигать положение равновесия катализаторы не могут, они в равной степени ускоряют как прямые, так и обратные реакции.
Любой катализатор активно взаимодействует с исходными реагентами, но его участие в процессе ограничивается только начальными стадиями превращений. В последующих стадиях он полностью регенерируется и может вновь взаимодействовать с молекулами реагирующих веществ. Этим и объясняется, что небольшого количества катализатора достаточно для получения очень больших количеств конечного продукта реакции. Факт снижения энергии активации химической реакции за счет образования промежуточных систем с участием катализатора является несомненным. Однако характер самого взаимодействия с катализатором может быть самым разнообразным.
Различают гомогенный и гетерогенный катализ. При гомогенном катализе катализатор и реагирующие вещества образуют однородную систему, например газовые смеси или жидкие растворы. При гетерогенном катализе катализатор чаще всего находится в твердой фазе, а реагирующие вещества — в газообразном или парообразном состоянии, т. е. в другой фазе. Возможны случаи, когда реагенты и катализатор — жидкости, но не смешивающиеся между собой. В нефтеперерабатывающей и нефтехимиче-
214
ской промышленности главным образом осуществляются гетерогенные каталитические процессы.
В последнее время для удобства рассмотрения механизма катализа принято все каталитические реакции делить на кислотно-основные и окислительно-восстановительные.
Кислотно-основными, или ионными, называются такие каталитические реакции, которые объясняются присоединением или отщеплением иона водорода (протона), а также реакции, при которых свободная пара электронов у реагирующих веществ или катализатора перемещается без разобщения электронов, образуя координационную связь в комплексном соединении. Отметим здесь же, что согласно теории Льюиса, кислотами называются соединения, молекулы которых способны присоединять электронную пару, т. е. включать ее в электронную оболочку одного из своих атомов.
Основаниями называются такие соединения, в молекуле которых имеется неподеленная пара электронов, способная внедряться в электронный слой кислоты. Это наиболее общая трактовка понятий кислоты и оснований. С такой точки зрения в группу кислот помимо обычных кислых веществ попадают и такие соединения, даже не содержащие водорода, как A1CU, FeCh, ZnCb, BF3. Все эти вещества — комплексообразующие катализаторы.
К кислотно-основным каталитическим реакциям можно отнести каталитический крекинг, полимеризацию, изомеризацию, гидратацию углеводородов и ряд других превращений.
Окислительно-восстановительными называются такие реакции, которые связаны с переходом электронов, т. е. протекают с разъединением электронной пары при разрыве валентной связи. К ним относятся многочисленные каталитические процессы окисления, гидрирования, дегидрирования и др.
Гомогенный катализ.Механизм гомогенного катализа хорошо объясняется теорией промежуточных химических соединений. По этой теории катализатор с реагирующим веществом образует неустойчивое реакционноспособное промежуточное соединение. Энергия активации этого процесса ниже энергии активации некаталитической основной реакции. В дальнейшем промежуточное соединение распадается или реагирует с новой молекулой исходного вещества, освобождая при этом катализатор в неизмененном виде. Эти превращения также характеризуются сравнительно малой энергией активации.
Гетерогенный катализ.При гетерогенном катализе все изменения и превращения веществ происходят на поверхности раздела твердой фазы катализатора и газовой (паровой) фазы реагирующих веществ и в значительной мере связаны с явлением сорбции. Процесс можно расчленить на следующие пять этапов:
1) движение (диффузия) реагирующих молекул к поверхности катализатора;
2) активированная адсорбция (хемоеорбция) реагирующих веществ на поверхности катализатора;
210
3) химическая реакция на поверхности катализатора;
4) десорбция (снятие с поверхности катализатора) продуктов реакции;
5) диффузия продуктов реакции в глубь газовой фазы.
Очевидно, скорость всего процесса определяется скоростью самого медленного этапа. Здесь различают два случая. Если диффузия исходных и конечных продуктов протекает быстрее, чем сама каталитическая реакция, то скорость процесса целиком зависит от состава и свойств поверхности катализатора. В этом случае говорят, что процесс протекает в кинетической области. Наоборот, если диффузия происходит медленнее, чем завершаются все превращения на поверхности катализатора,то общая скорость реакции будет определяться скоростью диффузии. В этом случае говорят, что процесс протекаете диффузионной области. Снижение энергии активации в гетерогенных каталитических реакциях достигается в результате сложных физико-химических процессов, характеризующих стадию активированной адсорбции (хемосорбции).
Хемосорбция — это образование достаточно прочного мономолекулярного слоя реагирующих веществ на поверхности катализатора. Хемосорбированные молекулы качественно отличны от молекул, находящихся в диффузионном слое. Хемосорбция протекает за счет валентных сил катализатора и по сути дела близка к настоящей химической реакции. Она характеризуется определенной энергией активации и ее следует отличать от обычной физической сорбции (адсорбции или абсорбции), которая не оказывает заметного влияния на прочность связей атомов в молекулах сорбированных веществ. Хемосорбция, наоборот, приводит к значительному ослаблению связей в реагирующих молекулах.
Различные теории катализа стремятся объяснить сущность хё-мосорбциТГ и характер изменений, происходящих в хемосорбиро-ванных молекулах. Интересно вспомнить, что еще Менделеев в '1886 г. писал о том, что на поверхности катализатора происходит деформация молекул реагирующих веществ, повышающая их реакционную способность. Именно этот принцип деформации молекул и положен в основу многих современных теорий катализа. В результате деформации молекулы поляризуются, связи в них ослабляются и иногда пязрываются, что приводит к появлению радикалов или атомов. Нередко в результате хеодрсорбции образуются нестойкие промежуточные соединения из молекул катализатора и сорбированного вещества. В этих случаях механизм гетерогенного катализа аналогичен механизму гомогенного катализа.
Активированная адсорбция молекул реагирующих веществ происходит не на всей свободной поверхности твердого катализатора, а только на так называемых активных центрах, где запас свободной энергии больше. Это могут быть острые углы, пики, различные неровности, ребра кристаллов, химически неоднородные участки и т. д. В целом, чем сильнее развита общая поверхность, тем больше на ней активных центров. Поэтому "првышеще
216
активности катализаторов часто связано с его высокой степенью измельчения и хорошо развитой пористой структурой.
Таким образом, общий принцип действия гетерогенных катализаторов заключается в том, что молекулы реагирующих веществ, сталкиваясь с активными центрами катализатора, могут образовать различные нестойкие промежуточные соединения (радикалы или ионы). От реакционной способности и природы этих соединений зависят и скорость протекающих реакций, и состав конечных продуктов.
В практике подбора и применения гетерогенных катализаторов необходимо учитывать их селективность, активность и срок службы.
Под селективностью, или избирательностью, катализатора понимают его способность ускорять только одну или несколько химических реакций определенного типа из числа, термодинамически вероятных в данных условиях для данного сырья. Это весьма важное свойство катализатора связано как с его составом, так и с условиями применения.
Активность катализатора характеризует его производительность. Чем активнее катализатор, тем меньше его нужно для превращения определенного количества исходных веществ в конечные продукты за единицу времени. Активность твердого катализатора зависит главным образом от состояния его поверхности. Катализаторы обычно применяются в виде таблеток, шариков или зерен небольших размеров. Для увеличения поверхности часто катализатор наносят на подложку (носитель), обладающую пористой поверхностью. В качестве носителей применяют активированный уголь, пемзу, кизельгур, окись алюминия, силикагель и искусственные цеолиты различных марок. Носитель повышает активность катализатора, придает ему механическую прочность и уменьшает, его расход. Активность многих катализаторов удается повысить добавлением небольшого количества так называемых промоторов, или активаторов. Действие активатороь может быть различным. Одни вещества увеличивают внутреннюю поверхность катализатора, т. е. воздействуют на его структуру и способствуют ее сохранению во время работы. Такие промоторы получили название структурных. Другие активаторы изменяют химический состав поверхности катализатора, увеличивают число активных центров. Такие активаторы получили название химических.
Активность катализаторов обычно определяют на лабораторных установках, где осуществляется тот технологический процесс, для которого предназначен Данный катализатор. Активность оценивают по выходу целевого продукта или* конверсии исходного сырья. Иногда испытуемый образец сравнивают с эталонным, активность которого известна. Активность катализаторов каталитического крекинга характеризуется так называемым индексом активности— выходом [в % (масс.) от сырья] фракции до 200 °С, полученной в результате каталитического крекинга эталонного сырья в стандартных условиях на лабораторной установке.
217
Катализатор после каждого элементарного акта реакции остается неизменным. Поэтому, казалось бы, он должен всегда работать неограниченно долго. Однако на практике срок службы катализатора всегда конечен, так как активность его со временем падает.
Продолжительность работы разных катализаторов различна. Так, например, алюмосиликатный катализатор для каталитического крекинга теряет активность через 10—15 мин, а вольфрамовые катализаторы деструктивной гидрогенизации работают 2— 3 года. Спад активности катализатора (дезактивация) часто называется его утомлением или старением. Причины этого явления различны. Чаще всего снижение активности катализатора наступает в результате отложения на его активных центрах продуктов реакции (кокса, смолы, парафинов и т. д.) или вследствие воздействия некоторых примесей в сырье, называемых каталитическими ядами. Последние особенно опасны, так как достаточно их ничтожного количества для полной дезактивации катализатора. Это заставляет весьма внимательно относиться к составу сырья. Часто для предотвращения отравления катализатора приходится исходное сырье подвергать дополнительной очистке. Действие каталитических ядов объясняется их необратимой адсорбцией на активных центрах катализатора.' Наиболее чувствительны-к ядам металлические катализаторы, содержащие Fe, Co, Ni, Pd, Ir, Pt, Cu, Ag. Часто встречаются, следующие каталитические яды: сероводород и другие сернистые соединения, окись углерода, азотистые основания, галогены, соединения фосфора, мышьяка и сурьмы, а также металлы (Ni, Fe, V), содержащиеся в тяжелом нефтяном сырье.
К причинам дезактивации катализаторов относятся также изменение структуры его поверхности, спекание и другие процессы, которые приводят к уменьшению поверхности или числа активных центров и могут быть вызваны условиями данного технологического процесса, главным-образом температурой.
Восстановление активности катализатора называется регенерацией. Способы регенерации различны. Углистые и смолистые отложения выжигают с поверхности катализатора в токе воздуха. Окисленные катализаторы восстанавливают действием водорода при определенной температуре. Катализаторы, содержащие редкие элементы (например, торий), регенерируют химическим путем. Редкий элемент извлекают из отработанного катализатора и вновь используют для приготовления свежих порций этого же катализатора.
В качестве катализаторов применяются разнообразные вещества: восстановленные металлы, окислы, кислоты, основания, соли, сульфиды металлов, некоторые органические соединения. Подбор катализаторов, изучение условий их изготовления и применения, пути регенерации и другие вопросы, связанные с катализом,—■ все это в настоящее время неотъемлемая часть нефтехимической технологии,
218
I.
I
я
1
1
•1
I
I -I
й щ Р"
ш
Р
в
Ы Р
■ п ■ It
Ш
■ ■ т Щ Щ
щ
ш
В!
ч
щ да
■
R И
л
4
i 1 i I
. i •I i
1
.] i
4 A
4
|
|
Хорошие результаты крекинга достигаются на предварительно гидроочищенном сырье. При гидроочистке значительно снижаются содержание серы, азота и металлов в сырье, а также его коксуемость. Крекинг подготовленного сырья приводит к уменьшению выхода кокса и газа и увеличению выхода бензина. Улучшается качество полученных продуктов: повышается октановое число бензина, содержание серы в жидких продуктах крекинга снижается настолько, что эти продукты не нуждаются в последующей очистке от серы. Положительный эффект дает также предварительное удаление смо-листо-асфальтеновых веществ из сырья методом деасфальтизации бензином или сжиженным пропаном.
| 440 460 480 Температура,°С Рис. 48. Зависимость выходов продуктов крекинга от температуры: / — бензин; 2 — газ: 3 — легкий газойль; 4 — кокс; 5—тяжелый газойль. |
Параметры процесса. Основными параметрами процесса каталитического крекинга являются температура, время контакта паров сырья с катализатором, определяемое объемной скоростью, и кратность циркуляции катализатора (при работе с движущимся катализатором).
Температура. В интервале температур 440—480°С образование бензиновых и дизельных фракций протекает достаточно быстро. С ростом температуры увеличивается и степень превращения сырья. Повышение температуры до 480—500 °С ведет к усилению газо- и коксообразова-ния и к снижению выхода' бензина. Октановое число бензина возрастает.
На рис. 48 приводится общий характер зависимости выхода продуктов крекинга от температуры процесса.
Давление. Процесс каталитического крекинга проводят под небольшим избыточным давлением 0,14—0,18 МПа.
Объемная скорость. Время контакта сырья и катализатора определяется объемной скоростью — отношением расхода жидкого сырья (в м3/ч) к объему катализатора (в м3), занимающего реакционную зону. Объемная скорость v0 [в м3/(м3-ч) или ч-1] определяется по формуле:
Va = Vc/vK
где vc, — расход сырья, м3/ч; vK — объем катализатора, м3.
Для пылевидного катализатора применяют величину массовой скорости подачи сырья [в кг/(кг-ч)], определяемую аналогично объемной.
Чем выше объемная скорость, тем ниже степень превращения. Повышение объемной скорости может быть скомпенсировано более высокой активностью катализатора, а также ростом температуры. При равных температуре и активности катализатора
8*
227
уменьшение объемной скорости приводит к увеличению степени превращения (рис. 49).
 Кратность циркуляции катализатора. Промышленные процессы каталитического крекинга осуществляются как на неподвижном, так и на непрерывно циркулирующем катализаторе. В последнем случае большое плияние на процесс оказывает соотношение количеств катализатора и сырья, подаваемых в реактор. Эта величина, называемая кратностью циркуляции катализатора, N (в кг/кг) определяется по формуле:
Кратность циркуляции катализатора. Промышленные процессы каталитического крекинга осуществляются как на неподвижном, так и на непрерывно циркулирующем катализаторе. В последнем случае большое плияние на процесс оказывает соотношение количеств катализатора и сырья, подаваемых в реактор. Эта величина, называемая кратностью циркуляции катализатора, N (в кг/кг) определяется по формуле:
N = RIB
| 500°С 450 400 |
| а?40 |
| 0 12 3 4 Объемная скорость, ч~ |
где R — количество катализатора, подаваемого в реактор, кг/ч; В — количество сырья, подаваемого в реактор, кг/ч.
| Рис. 49. Зависимость степени превращения сырья от объемной скорости при работе на цеолитном катализаторе. где R и В выражаются в м3/ч. Между массовой и объемной кратностью циркуляции катализатора существует соотношение-: |
Наряду с массовой применяют объемную кратность циркуляции катализатора N0:
N0 = R/B
N0
тЙг
N
где рс — плотность сырья при 20 °С, кг/м3; pi-aT—плотность катализатора, кг/м3.
Увеличение кратности циркуляции ведет к сокращению -продолжительности пребывания катализатора в зоне реакции. Количество кокса на каждой частице уменьшается. Средняя активность катализатора возрастает, а это способствует увеличению степени превращения, т.е. повышению выхода газа, бензина и кокса. В целом абсолютное количество кокса возрастает, но оно откладывается на большем числе частиц.
Изменением кратности циркуляции катализатора можно регулировать количество теплоты, вносимой в реактор, степень превращения сырья, степень закоксованносги катализатора на выходе из реактора. С экономической точки зрения повышение кратности циркуляции приводит к увеличению размеров регенератора и росту эксплуатационных расходов на перемещение катализатора. Количество кокса на входе в регенератор не должно превышать 0,8— 1%(масс), остаточное содержание кокса после регенерации не более 0,25% (масс.) в расчете на катализатор. Для поддержания рабочей активности приходится выводить из системы часть катализатора и заменять его свежим. Расход катализатора -—2 2,3 ю на 1000 кг сырья.
228
 Тепловой эффект. Тепловой эффект каталитического крекинга является суммой тепловых эффектов отдельных реакций процесса и зависит от степени превращения. При степени превращения 80—90% отрицательный тепловой эффект реакции 230— 290 кДж/кг сырья (рис. 50).
Тепловой эффект. Тепловой эффект каталитического крекинга является суммой тепловых эффектов отдельных реакций процесса и зависит от степени превращения. При степени превращения 80—90% отрицательный тепловой эффект реакции 230— 290 кДж/кг сырья (рис. 50).
Технологический режими материальный баланс процесса. Примерный технологический режим и материальный баланс процесса при переработке вакуумного газойля фракции 350—500 °С на шариковом и пылевидном катализаторе цеокар-2 приводится в табл. 10.
| 0,2 0,4 0,6 0,8 Степень превращения |
| 1,0 |
| Рис. 50. Зависимость теплового эффекта каталитического крекинга вакуумного газойля ромашкинской нефти от степени превращения сырья (выход фракции до 360 °С) н,1 катализаторе типа АШНЦ-3. |
Объемная скорость обычно составляет от 1,5 для шарикового и до 3 ч-1 для микросферического цеолит-содержащего катализатора. Кратность циркуляции изменяется в более широких пределах: от 2,5—4 на шариковом до 7,5—10 на микросферическом цеолитсодержащем катализаторе. На более легком сырье кратность циркуляции выше, чем на тяжелом сырье.
Качество продуктов каталитического крекинга.Углеводородные газы каталитического крекинга содержат не менее 75—80% (масс.) смеси пропан-пропиленов, бутан-бутиленов и пен-тан-амиленов. Содержание изомерных соединений достигает 25— 40%(масс). Это делает газы каталитического крекинга ценным сырьем для нефтехимических процессов.
ТА6ЛИЦА 10
Дата добавления: 2018-04-04; просмотров: 1413; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!