Основные этапы химического анализа.
Этап 1. Постановка аналитической задачи. какой материал подвергается анализу и что именно в нем следует определять
Этап 2. Выбор метода и методики анализа. учитывается: поставленная аналитическая задача,размеры объекта, примерное содержание определяемых компонентов
В качественном анализе пределом обнаружения называется наименьшее количество вещества, которое с уверенностью обнаруживается данной реакцией. Аналитическая реакция тем чувствительнее, чем ниже предел обнаружения. Предел обнаружениязависит от концентрации реагентов, присутствия посторонних ионов электролитов или мешающих веществ, среды и температуры растворов. Его можно повысить с помощью приемов концентрирования, понижения растворимости осадка.
В качественном анализе пределом обнаружения называется наименьшее количество вещества, которое с уверенностью обнаруживается данной реакцией. Аналитическая реакция тем чувствительнее, чем ниже предел обнаружения. Предел обнаружениязависит от концентрации реагентов, присутствия посторонних ионов электролитов или мешающих веществ, среды и температуры растворов. Его можно повысить с помощью приемов концентрирования, понижения растворимости осадка.
5.Дробный и Систематический анализАналитическим сигналом может быть цвет и его изменение, запах, выделение газообразных продуктов, окрашивание пламени, образование люминесцирующих соединений, выпадение или растворение осадка.
|
|
|
В случае образования осадка, кроме самого факта выпадения, аналитическим сигналом может служить его цвет, форма (кристаллическая или аморфная), а также характерная форма кристаллов.
Аналитические реакции, согласно рекомендации ИЮПАК, подразделяют на специфические и избирательные (селективные) методы, реакции и реагенты.
Специфическими называют те методы, реакции или реагенты, с помощью которых в данных условиях можно обнаружить только одно вещество; избирательными — методы, реакции и реагенты, позволяющие обнаружить небольшое число веществ.
Специфические реагенты и реакции позволяют обнаружить данное вещество или ион в присутствии других веществ или ионов.
Селективные реагенты и реакции позволяют обнаружить несколько веществ или ионов. Таких реагентов и реакций известно существенно больше, чем специфических.
Избирательность достигается правильным выбором и установлением соответствующих условий реакции. К факторам, определяющим условия протекания реакции, относят рН, температуру, концентрации открываемого и посторонних ионов, природу растворителя (вода, органические или водно-органические среды). Реакции или реагент можно сделать более избирательными или даже специфичными варьированием рН, концентраций, маскированием, изменением степени окисления элементов, температуры.
|
|
|
Реагенты по их избирательности можно разделить на три группы:
1. Специфические реагенты — например: крахмал для обнаружения I2; NaOH для открытия 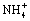 .
.
2. Избирательные (селективные) реагенты — например: диметилглиоксим в аммиачной среде реагирует с Fe(II), Co(II), Ni(II), Zr(IV) и Th(IV).
3. Групповые реагенты — например: HCl осаждает нерастворимые хлориды Ag(I), Hg(I), Tl(I), Pb(II).
Групповые и селективные реагенты, образующие малорастворимые соединения с ионами, используют при разделении катионов на аналитические группы. Ионы, осаждаемые данным групповым реагентом, образуют аналитическую группу ионов. Используемый групповой реагент и условия осаждения определяют состав аналитической группы ионов.
Анализ смеси ионов — трудная аналитическая задача, поскольку посторонние ионы могут препятствовать открытию интересующего нас иона. Такие ионы называют мешающими. Помехи со стороны сопутствующих ионов начинают проявляться при определенном соотношении открываемых и мешающих ионов и усиливаются с увеличением концентрации последних. Причинами мешающеговлияния при открытии ионов могут быть:
|
|
|
1. Образование (мешающими ионами) при действии реагента соединений, аналогичных аналитической форме открываемого иона, и генерирование ими такого же сигнала, что и аналитический сигнал открываемого иона.
2. Образование (мешающими ионами) соединений, препятствующих наблюдению аналитического сигнала.
3. Вероятность проведения аналитической реакции открытия невелика, например, в результате связывания реагента мешающими ионами.
Для устранения мешающего влияния сопутствующих ионов используют два пути:
· избирательное распределение компонентов анализируемой системы между двумя разделяющимися фазами (методы разделения или отделения). В практике качественного анализа наибольшие значения имеют осаждение, экстракция и хроматография;
· маскирование мешающих ионов. Для этого используют химические реакции, протекающие в той же фазе, что и реакции обнаружения, и приводящие к уменьшению концентрации мешающего иона или реагента. Для маскирования и демаскирования применяют реакции комплексообразования, кислотно-основные и окислительно-восстановительные.
Реальные объекты всегда многокомпонентны, часто и многофазны. Для открытия каждого иона необходимо:
|
|
|
· создать условия протекания частной реакции;
· устранить мешающее влияние сопутствующих компонентов;
· зарегистрировать аналитический сигнал.
Возможны два пути решения этой задачи: проведение систематического качественного химического анализа или дробное обнаружение ионов.
Последовательное разделение ионов на отдельные аналитические группы методом осаждения групповыми реагентами (или иным образом) называют систематическим ходом анализа.
Метод дробного обнаружения основан на применении специфических и селективных реагентов без разделения на группы. Мешающее влияние сопутствующих ионов устраняют, используя приемы маскирования, а также применяя более селективные или специфические реагенты.
Возможности дробного открытия ионов были существенно расширены благодаря применению органических реагентов, значительная часть которых специально синтезирована для повышения селективности реакций обнаружения. Широкие возможности органических реагентов связаны, во-первых, с их многообразием и, следовательно, возможностью выбора подходящего реагента, а во-вторых, с разнообразием свойств образуемых ими комплексов, таких, как устойчивость, растворимость, летучесть, окраска, окислительно-восстановительные свойства. Метод дробного определения может быть применен к анализу аналитических групп ионов, выделенных групповыми реагентами.
Таким образом, процедура качественного химического анализа представляет собой последовательное отделение аналитических групп с дальнейшим открытием входящих в них ионов систематическим или дробным методами. В ходе выполнения анализа как систематическим, так и дробным методами аналитик управляет поведением ионов в растворе, прежде всего их концентрациями. Такое управление возможно на основе равновесных реакций путем смещения равновесий. В распоряжении аналитика два типа равновесных процессов — гомогенные и гетерогенные равновесия. Гомогенные равновесия — это диссоциация — ассоциация, окисление — восстановление, гидролиз, нейтрализация, комплексообразование. Количественное описание этих равновесий основано на законе действующих масс и уравнении Нернста для окислительно-восстановительного потенциала системы. К гетерогенным равновесиям относятся, прежде всего, растворение и осаждение осадков, экстракционное распределение между двумя жидкими фазами и хроматографические процессы. Расчеты положения гетерогенного равновесия возможны на основе констант межфазных распределений, в первую очередь правила произведения растворимости.
Характеристика чувствительности аналитической реакции выражается в том, что аналитические реагенты и аналитические реакции позволяют обнаруживать определяемое вещество в пробе, если его содержание превышает некоторый минимальный предел. Если концентрация определяемого вещества ниже этого предела, то и концентрация аналитической формы окажется настолько незначительной, что невозможно будет зарегистрировать аналитический сигнал.
Предел обнаружения — минимальная концентрация или минимальное количество вещества, которое может быть обнаружено данным методом с какой-то допустимой погрешностью. Его обозначают сminP, где Р — доверительная вероятность. Чувствительность характеризует изменение сигнала с изменением концентрации и выражается коэффициентом чувствительности, который численно равен тангенсу угла наклона линейной зависимости аналитического сигнала от концентрации.
Предел обнаружения в качественном анализе традиционно называли открываемым минимумом. В настоящее время в качественном анализе используется большое число реагентов и частных реакций с низкими пределами обнаружения. Обычно для открытия ионов применяют реакции с пределом обнаружения 10–7 г (0,1 мкг) в 1 мл раствора. Физические методы позволяют открыть элементы в твердых образцах с пределом обнаружения менее 10–15 г.
В химических методах качественного анализа предел обнаружения может быть существенно понижен при использовании органических реагентов, особенно в случае образования открываемым ионом смешано-лигандных комплексов. Для этих же целей используют ряд приемов практического проведения реакции — таких, как микрокристаллоскопический анализ, капельный анализ, флотация, жидкостная экстракция, метод умножения реакций, каталитические и люминесцентные реакции, реакции на носителях.
Предел обнаружения, наряду с избирательностью, является важнейшей характеристикой реакции и метода анализа.
Систематический анализ основан на последовательном выделении из растворовотдельных групп ионов, на выделении отдельных ионов из подгрупп. Выделенные ионыопределяют при помощи соответствующих реакций. На исследование берутнавески исследуемого объекта и операции (минерализация, осаждение, растворение, фильтрование)Дробный метод основан на применении реакций, с помощью которых в любой последовательности можно обнаружить искомые ионы в отдельных небольших порциях исследуемого раствора. Для обнаружения соответствующих ионов необходимо применять специфические реактивы, позволяющие обнаружить искомый ион в присутствии посторонних ионов. Обнаружение производится в два этапа. Вначале устраняют влияние мешающих ионов с помощью соответствующих реактивов, а затем прибавляют реактив, дающий окраску или осадок с искомым ионом. 5.Дробный и Систематический анализСистематический анализ основан на последовательном выделении из растворовотдельных групп ионов, на выделении отдельных ионов из подгрупп. Выделенные ионыопределяют при помощи соответствующих реакций. На исследование берутнавески исследуемого объекта и операции (минерализация, осаждение, растворение, фильтрование)Дробный метод основан на применении реакций, с помощью которых в любой последовательности можно обнаружить искомые ионы в отдельных небольших порциях исследуемого раствора. Для обнаружения соответствующих ионов необходимо применять специфические реактивы, позволяющие обнаружить искомый ион в присутствии посторонних ионов. Обнаружение производится в два этапа. Вначале устраняют влияние мешающих ионов с помощью соответствующих реактивов, а затем прибавляют реактив, дающий окраску или осадок с искомым ионом.
6. Разделение катионов на аналитические группы. При анализе образцов в которых несколько видов катионов их взаимно мешающее действие не позволяет воспользоваться дробным методом анализа. Пользуются системным анализом(разделяют ионы групповым реагентом). Теоретически базой для классификации катионов может служить ожжет служить Табл. Менделеева, которая характеризуется четкой аналогией совокупных свойств внутри каждой группы элементов. 1 гр. (Na,K,NH4) группового реагента нет 2 гр. (Ag,Hg,Pb,)- групповой реагент HCl
3 гр. (Ca, Ba, Pb)- групповой реагент H2SO4
4 гр. (AlCrZnAsSn)- групповой реагент NaOH 5 гр. (BiMgMnFe)- групповой реагент NaOH 6 гр. (CoCuNiCdHg) – групп. реагент NH4OH Существует несколько схем классификации катионов по аналитическим группам. Одной из наиболее распространенных является классификация, основанная на свойствах хлоридов, сульфидов и карбонатов. При разделении по так называемому сероводородному методу систематический ход анализа заключается в следующем.
· 1. Раствором хлороводородной кислоты осаждают нерастворимые хлориды: AgCl, Hg2Cl2H РЬС12. Таким образом, катионы Ag+, Hg2+, Pb2+ составляют так называемую 5-ю аналитическую группу.
· 2. Раствор, имеющий кислую реакцию, обрабатывают сероводородом. При этом в осадок переходят катионы меди(И), кадмия(И), ртути(И), висму- та(Ш), мышьяка, сурьмы и олова, сульфиды которых нерастворимы в разбавленных кислотах. Эти катионы образуют 4-ю аналитическую группу.
· 3. Отделив раствор от осадка, его нейтрализуют до слабощелочной реакции и обрабатывают сульфидом аммония, который осаждает катионы железа, цинка, марганца(И), никеля(П), кобальта(П), хрома(Ш), алюмипия(Ш) и ряда других элементов. Эти катионы составляют 3-ю аналитическую группу.
· 4. После отделения осадка, содержащего катионы 3-й группы, катионы щелочноземельных элементов (кроме магния) осаждают из раствора в виде карбонатов. Эта группа называется 2-й аналитической группой.
· 5. После отделения карбонатов 2-й группы в растворе остаются катионы щелочных металлов, магния и аммония, которые не имеют группового реагента и составляют 1-ю аналитическую группу.
Итак, в основу классификации катионов положено различие в растворимости образуемых ими соединений, позволяющее отделять одни группы ионов от других (табл. 3.1).
Классификация катионов
| Сульфиды, растворимые в воде | Сульфиды, нерастворимые в воде (гидроксиды, образующиеся при действии группового реагента) | |||
| Карбонаты, растворимые в воде | Карбонаты, нерастворимые в воде | Сульфиды (гидроксиды), растворимые в разбавленных кислотах | Сульфиды, нерастворимые в разбавленных кислотах | |
| 1-я группа | 2-я группа | 3-я группа | 4-я группа | 5-я группа |
| К Na NH/, Mg2 (MgC03растворим только в присутствии солей аммония) | Ва2*, Sr2*, Са2‘ | А13*, Cr3*, Fe3*, Fe2*, Mg2*, Zn2*, Co2*, Ni2* | а) первая подгруппа: сульфиды не образуют тиосолей при действии Na2S; в) вторая подгруппа: сульфиды растворяются в Na2S с образованием тиосолей | Хлориды нерастворимы в воде и разбавленных кислотах (Ag Hg2 Pb2+) |
| Группового реагента не имеют | Групповой реагент (NH4)2C03 | Групповой реагент (NH4)2S | Групповой реагент H2S + НС1 | Групповой реагент НС1 |
Применение групповых реагентов позволяет задачу анализа свести к определению катионов в пределах группы, если она присутствует в растворе.
Неметаллы в воде содержатся обычно в виде анионов соответствующих кислот. Исключение составляет азот, который может существовать как в форме анионов N03 и NO^, гак и в форме катиона NHJ, и углерод, присутствующий в виде неорганических анионов и органических веществ. Кроме того, следует иметь в виду, что некоторые металлы также могут образовывать анионы, например, такие, как СгОл_, [А1(ОН)6]л , и ряд других.
Анионы принято делить на три аналитические группы в зависимости от растворимости соответствующих солей бария и серебра. Групповыми реагентами служат хлорид бария и нитрат серебра (табл. 3.2).
Таблица 3.2
Классификация анионов
| Номер аналитической группы | Анионы, составляющие группу | Характеристика группы | Групповой реагент |
| 1 | SO2-, СО2-, Р043-, SiO2-, AsO3-, AsO3-, CrO2-, Cr2()2 , F идр. | Соли бария малорастворимы в воде, но растворимы в разбавленных кислотах (кроме BaS04) | Раствор ВаС12 в нейтральной или слабощелочной среде |
| 2 | Cl , Br , Г, S2 и др. | Соли серебра нерастворимы в воде и в азотной кислоте | Раствор нитрата серебра в присутствии азотной кислоты |
| 3 | N03,N02,CH3C00 , и др. | Соли бария и серебра растворимы в воде | Группового реаген |
7.Закон действия масс. Теоретические основа качественного анализа в 1864г Гульдберг и Вааге открыли закон действующих масс. Он является теоретической основой многих методов анализа, поскольку устангавливает связь между скоростью и молярными концентрациями веществ, участвующих в обратимой химияческойреакции.СКОРОСТЬ ХИМИЧЕСКИХ РЕАКЦИЙ ПРОПОРЦИОНАЛЬНА ПРОИЗВЕДЕНИЮ КОНЦЕНТРАЦИЙ РЕАГИРУЮЩИХ ВЕЩЕСТВ. А+б=с+д v=k1(а)(б) v2=k2(с)(д) K=v/v2=(с)(д) /(а)(б) K-константа равновесия(поскольку к1 и к2 для участвующих в реакции веществ при неизменной температуре – постоянные величины, то и отношение их – величина постоянная, К) это уравнение является следствием закона ПРИ ОБРАТИМЫХ РЕАКЦИЯХ РАВНОВЕСИЕ НАСТУПАЕТ, КОГДА ОТНОШЕНИЕ ПРОИЗВЕДЕНИЯ КОНЦЕНТРАЦИЙ ОБРАЗУЮЩИХСЯ ВЕЩЕСТВ К ПРОИЗВЕДЕНИЮ КОНЦЕНТРАЦИЙ ВСТУПАЮЩИХ В РЕАКЦИЮ ВЕЩЕСТВ СТАНОВИТСЯ РАВНЫМ НЕКОТОРОЙ ПОСТОЯННОЙ ДЛЯ ДАННОЙ ХИМИЧЕСКОЙ РЕАКЦИИ ВЕЛИЧИНЕ, НАЗЫВАЮЩЕЙСЯ КОНСТАНТОЙ ХИМИЧЕСКОГО РАВНОВЕСИЯ
Зависимость скорости реакции от концентрации реагирующих веществ, сформулированная Гульдбергом Като Максимилианом (1836 – 1902) – норвежским физикохимиком и математиком и Петером Вааге (1833 – 1900) норвежским физикохимиком и минералогом в 1864 – 1867 г. г. получила название закона действующих масс: скорость химической реакции прямо пропорциональна произведению молярных концентраций реагирующих веществ, возведенных в степени их стехиометрических коэффициентов.
Рассмотрим обратимую реакцию общего вида
Экспериментальные исследования показывают, что в состоянии равновесия выполняется следующее соотношение:
(квадратные скобки означают концентрацию). Приведенное соотношение представляет собой математическое выражение закона действующих масс, или закона химического равновесия, согласно которому в состоянии химического равновесия при определенной температуре произведение концентраций продуктов реакции в степенях, показатели которых равны соответствующим коэффициентам в стехиометрическом уравнении реакции, деленное на аналогичное произведение концентраций реагентов в соответствующих степенях, представляет собой постоянную величину. Эта постоянная Кс называется константой равновесия. Выражение константы равновесия через концентрации продуктов и реагентов характерно для реакций в растворах.
Отметим, что правая часть выражения для константы равновесия содержит только концентрации растворенных веществ. Она не должна включать никаких членов, относящихся к участвующим в реакции чистым твердым веществам, чистым жидкостям, растворителям, так как эти члены постоянны.
Для реакций с участием газов константа равновесия выражается через парциальные давления газов, а не через их концентрации. В этом случае константу равновесия обозначают символом Кр.
Концентрацию газа можно выразить через его давление при помощи уравнения состояния идеального газа (см. разд. 3.1): pV=nRT
8. Состояние сильных электролитов в растворах. В реальных растворах все ионы окружены ионной атмосферой состоящей из противоположно заряженных ионов. В результате этого их подвижность уменьшается и они движутся медленнее и изменяется их электропроводность поэтому вводят понятие активности определяющей действие концентрации в растворе с учетом ионного взаимодействия. Подстановка активностейвместо концентраций в уравнения, определяющие условия фазовых, химических или электрохимических равновесий для идеальных растворов, делает эти уравнения применимыми к реальным растворам. Наряду с активностью пользуются коэффициентом активности, равным отношению активностик концентрации. Ионы не могут выделиться из раствора порознь, а лишь в таком сочетании, при котором сохраняется электронейтральность раствора. Поэтому вводят в рассмотрение активность сильного электролита как целого. Эта величина принимается по определению равной произведению активностейионов, на которые молекулараспадается при электролитической диссоциации. За коэффициент активности сильного электролита принимают среднее геометрическое из коэффициентов активностей его ионов; коэффициенты активности ионов считаются равными отношениям активностей к концентрациям(так же, как в случае неэлектролитов).
Константадиссоциаци. - константа равновесия реакциидиссоциации, степень диссоциации (доля продиссоциировавших частиц) - отношение числа продиссоциировавших частиц к общему числу частиц, введенных в систему
.КОНСТАНТА РАВНОВЕСИЯ-величина, определяющая для данной хим. р-ции соотношение между термодинамич. активностями исходных в-в и продуктов в состоянии хим. равновесия. реакции АВ " A+ + B-
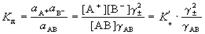
Проводники, прохождение через которые электрического тока вызывает перемещение вещества в виде ионов (ионная проводимость) и химические превращения (электрохимические реакции), называются электролитами. Это могут быть индивидуальные вещества или растворы.
Упрощенная формулировка: электролиты - это вещества, способные распадаться на ионы в растворах. Правда, такая формулировка является менее общей и не охватывает твердые электролиты и расплавы электролитов.
Термин «ион» впервые ввел английский физик М. Фарадей (1791-1867).
Раствор - это гомогенная смесь двух или нескольких веществ, способная непрерывно изменять свои свойства. Растворы бывают жидкие и твердые. В аналитике используют в основном жидкие растворы.
В соответствии с теорией электролитической диссоциации (1883-1887) шведского ученого (1859-1927), который за создание этой теории был удостоен в 1902 г. Нобелевской премии, электролиты в растворах распадаются (диссоциируют) на ионы вследствие взаимодействия с молекулами растворителя.
Количественно ионизация (диссоциация на ионы) электролита в растворе характеризуется степенью диссоциации (ионизации) α, равной отношению числа продиссоциировавших молекул пдисс к исходному числу молекул писх:
α = пдисс/ писх.
Степень диссоциации (ионизации) α численно выражается либо в долях единицы, либо в процентах. Если α = 1 (т. е. 100%), то все исходные частицы в растворе распались на ионы (пдисс = писх); если α < 1 (т. е. меньше 100 %), то не все исходные частицы распались на ионы, а только часть их (пдисс < писх).
По способности к диссоциации электролиты разделяют на сильные (неассоциированные) и слабые (ассоциированные).
Сильные (неассоциированные) электролиты в не слишком концентрированных растворах распадаются на ионы практически полностью. Это - большинство солей, сильные кислоты, сильные основания. Например, в водных растворах хлорида натрия NaCl, хлороводородной кислоты НС1, гидроксида натрия NaOH диссоциация на ионы осуществляется нацело:
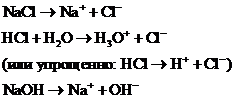
Для сильных электролитов степень их ионизации 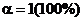 .
.
В концентрированных растворах сильные электролиты частично, хотя обычно в очень небольшой степени, ассоциированы.
Слабые (ассоциированные) электролиты в растворах диссоциированы лишь частично. Это -слабые кислоты, слабые основания, комплексные соединения (их внутренняя сфера), некоторые соли ртути(ІІ), например, HgCl2 Hg(CN)2. Так, в водных растворах уксусная кислота и аммиакдиссоциируют лишь частично:

Аналогично тетрахлороплатинат(ІІ)-ион диссоциирует также частично:
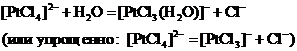
Обычно степень диссоциации α для слабых электролитов очень мала ( 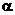
 1) и уменьшается с ростом концентрации раствора.
1) и уменьшается с ростом концентрации раствора.
Понятие сильные и слабые электролиты ˗ условно, поскольку одно и то же вещество в одних растворителях может быть сильным, а в других ˗ слабым электролитом. Например, такие кислоты, как хлорная 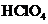 и хлороводородная НС1, являются сильными электролитами (диссоциируют нацело) в воде и в жидком аммиаке, однако в безводной уксусной кислоте они оказываются слабыми электролитами и распадаются на ионы лишь в незначительной степени. Напротив, уксусная кислота СН3СООН - слабый электролит в водных растворах, но сильный - в жидком аммиаке.
и хлороводородная НС1, являются сильными электролитами (диссоциируют нацело) в воде и в жидком аммиаке, однако в безводной уксусной кислоте они оказываются слабыми электролитами и распадаются на ионы лишь в незначительной степени. Напротив, уксусная кислота СН3СООН - слабый электролит в водных растворах, но сильный - в жидком аммиаке.
По Аррениусу сильные электролиты также диссоциированы на ионы неполностью. Строго говоря, теория Аррениуса неприменима к растворам сильных электролитов. До Аррениуса большинство ученых считало, что ионы возникают только под воздействием внешнего электрического поля при электролизе. Правда, прибалтийский ученый К. (1785 ˗ 1822) в 1818 г. предположил, что распад молекул на ионы происходит в результате процесса растворения, а не вследствие воздействия внешнего электрического поля. Ту же точку зрения разделяли немецкий физик Р. Клаузиус в 1857 г. и русский химик (1851 ˗ 1896) в 1881 г. Однако только после создания теории электролитической диссоциации Аррениуса общепринятым стало представление о том, что электролиты распадаются на ионы уже при растворении, в отсутствии внешнего электрического поля.
Простейшее (но далеко не исчерпывающее) объяснение причин электролитической диссоциации заключается в следующем.
В вакууме силам притяжения между катионом и анионом не мешают никакие посторонние частицы. В любой другой среде энергия взаимодействия между электрическими зарядами уменьшается, что характеризуется диэлектрической проницаемостью 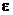 ( - число, показывающее, во сколько раз уменьшается энергия взаимодействия между электрическими зарядами в данной среде по сравнению с вакуумом).
( - число, показывающее, во сколько раз уменьшается энергия взаимодействия между электрическими зарядами в данной среде по сравнению с вакуумом).
Пусть некоторый электролит КА, состоящий из катиона 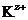 и аниона
и аниона 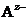 , распадается на ионы:
, распадается на ионы:
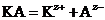
В соответствии с законом Кулона в вакууме энергия электростатического Взаимодействия Еточечных электрических зарядов и равна:
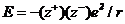
где е ˗ единичный электрический заряд; r ˗ расстояние между центрами катиона и аниона . В среде с  энергия взаимодействия между теми же электрическими зарядами ибудет другой — в раз меньше:
энергия взаимодействия между теми же электрическими зарядами ибудет другой — в раз меньше:
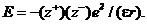
Например, для воды при 18°С диэлектрическая проницаемость = 81, т. е. энергия электростатического взаимодействия между электрическими зарядами в водной среде уменьшается в 81 раз по сравнению с вакуумом. Для электролитов в водных растворах это означает увеличение их способности к ионизации. Полагают, что значительную роль при этом играют процессы сольватации: молекулы растворителя, например, воды, окружая катион и анион, создают сольватные (гидратные ˗ в случае воды) Оболочки вокруг ионов и как бы «растаскивают» их.
9.
Теории кислот и оснований (Аррениуса, Льюиса, Бренстеда-Лоури) одной из первых теорий, объясняющих природу кислот и оснований явилась теория электролитич.диссоциации Аррениуса-Оствальда. Согласно ей кислотой является электронейтральноеве-во, которое при диссоциации в воде в качестве катиона обрзует только ионы водорода Н+. НА↔Н++А- А основанием явл.ве-во, кот.при диссоциации образует в воде в кач.аниона только гидроксид-ионы NaOH→Na++OH- Но так как возникали некот.трудности (некот.ве-ва проявляли св-ва солей, кислот и т.д), то пришла на смену протолитическая теория кислот и оснований- Бренстеда-Лоури, согласно которой кислотой явл.любоеве-во, способное отдавать протон, а основанием способное принимать протон. Многие соли в водных р-рах ведут себя как кислоты, хотя Н+ в их составе нет. Эти факты были объяснены в теории Льюиса, но она не нашла широкого применения. 5 Протолитическая теория: анализ роли растворителя. Классификация растворителей. Теория Бренстеда-Лоури, согласно которой кислотой явл.любоеве-во, способное отдавать протон, а основанием способное принимать протон. Растворитель явл.не только явл.средой, но и участником процесса. Растворители: 1. протонные-могут отдавать или принимать протон. · Кислотные- спос-ть к отдаче протона, основные-спос-ть присоединять протон, амфотерные. 2. апротонные – не обладают донорно-акцепторными св-ми по отношении к протону. Если ве-во взято в кач.растворителя, то в зависимости от того, какая тенденция отдача или присоединение протона у него преобладает, то оно может различным образом влиять на ионизацию растворенного ве-ва По влиянию на кислотно-основные св-ва%: невелирующие и дифференцирующие.
10 Буферные растворырастворы с определённой устойчивой концентрацией водородных ионов; смесь слабой кислоты и её соли (напр., СН3СООН и CH3COONa) или слабого основания и его соли (NH3 и NH4CI). Величина рНбуферного раствора мало изменяется при добавлении небольших количеств свободной сильной кислоты или щёлочи, при разбавлении или концентрировании. Буферные растворы широко используют в различных химических исследованиях. Для определения пределов действия Б.р. вводится понятие буферная емкость, измеряемая количеством сильной кислоты или основания (в г-экв), которое надо добавить к 1 л Б.р., чтобы сместить рН на единицу.
10. Буферные растворы Сущ-т р-ры при + к которым небольш.кол-ва кислоты, щелочи или при их разбавлении рн изменяется незначительно- буферные р-ры. А указанная их способность поддерживать рн практически постоянной наз-ся буферным действием. Буф.спос-тью обладают смеси след.типа: 1.смесь слабой к-ты и соли с одноименным анионом. CH3COOH+CH3COONa
2. смесь слабого основания и соли с одноименным катионом NH3+NH4Cl
3. смесь слабой многоосновной к-ты различной степени замещенности NaH2PO4+Na2KPO4
Буферное действие р-ров хар-ся буферной емкостью. Это то мин.кол-во сильной к-ты или щелочи, кот.необходимо добавить к 1 л. Буферного р-ра, чтобы его рН изменилась на еденицу. рН буф.р-ров зависит от конст. диссоциации слабой к-ты или основания и от компонентов буф.р-ра.
[H+]= Ккисл*(Скисл./Ссоли)
[ОH-]= Косн.*(Сосн../Ссоли)
(т.е. соли, образованной сильной кислотой и катионом этого основания). Например: NH4OH и NH4Cl –Уравнение буферной системы рассчитывается по формуле Гендерсона-Гассельбаха:
рН = рК + ℓg 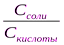 , pOH = pK + ℓg
, pOH = pK + ℓg  ,
,
где рК = -ℓg КД.
С – молярная или эквивалентная концентрация электролита (C = V N)
Механизм действия буферных растворов
Рассмотрим его на примере ацетатного буфера: СН3СООН + СН3СООNaПри добавлении небольшого количества хлороводородной кислоты, ионы Н+связываются с имеющимся в растворе сопряженным основанием СН3СОО-в слабый электролит СН3СООН.
CH3COO‾ +H+↔CH3COOH(1)
Из уравнения (1) видно, что сильная кислота НС1 заменяется эквивалентным количеством слабой кислоты СН3СООН. Количество СН3СООН увеличивается и по закону разбавления В. Оствальда степень диссоциации уменьшается. В результате этого концентрация ионов Н+ в буфере увеличивается, но очень незначительно. рН сохраняется постоянным.
При добавлении кислоты к буферу рН определяется по формуле:
рН = рК + ℓg 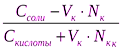
При добавлении к буферу небольшого количества щелочи протекает реакция её с СН3СООН. Молекулы уксусной кислоты будут реагировать с гидроксид-ионами с образованием Н2О и СН3СОО ‾:
CH3COOН +OH ‾↔CH3COO‾ +H2O(2)
В результате этого щелочь заменяется эквивалентным количеством слабоосновной соли CH3COONa. Количество СН3СООН убывает и по закону разбавления В. Оствальда степень диссоциации увеличивается за счет потенциальной кислотности оставшихся недиссоциированных молекул СН3СООН. Следовательно, концентрация ионов Н+практически не изменяется. рН остаётся постоянным.
При добавлении щелочи рН определяется по формуле:
рН = рК + ℓg 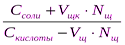
При разбавлении буфера рН также не меняется, т.к. константа диссоциации и соотношение компонентов при этом остаются неизменными.
Таким образом, рН буфера зависит от: константы диссоциации и соотношения концентрации компонентов. Чем эти величины больше, тем больше рН буфера. рН буфера будет наибольшим при соотношении компонентов равным единице.
Для количественной характеристики буфера вводится понятие буферной ёмкости.
Буферная ёмкость
Это способность буферной системы противодействовать изменению рН среды.Интервал значений рН, выше и ниже которого буферное действие прекращается, называется зоной буферного действия.Она равна рН = рК ± 1Буферная ёмкость (В) выражается количеством моль-эквивалентов сильной кислоты или щелочи, которое следует добавить к одному литру буфера, чтобы сместить рН на единицу.
В = 
В – буферная ёмкость,
nЭ– количество моль-эквивалента сильной кислоты или щелочи,
рНН – начальное значение рН ( до добавления кислоты или щелочи)
рНК– конечное значение рН (после добавления кислоты или щелочи)
ΔрН – изменение рН.
буферная ёмкость рассчитывается по формуле:
В = 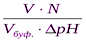
V – объём кислоты или щелочи,
N – эквивалентная концентрация кислоты или щелочи,
Vбуф.- объём буферного раствора,
Δ рН – изменение рН.
Буферная ёмкость зависит от концентрации электролитов и соотношения компонентов буфера. Наибольшей буферной ёмкостью обладают растворы с большей концентрацией компонентов и соотношением компонентов, равным единице.В организме человека действуют белковый, гемоглобиновый, фосфатный и бикарбонатный буферы.
Типы комплексных соединений, используемых в аналитической химии. Их свойства. Комплексообразование с монодентатными и полидентатнымилигандами: строение комплексных соединений, равновесия в растворах комплексных соединений, константы устойчивости комплексных ионов.
Компл.соед.в аналитической химии. Кач.анализ катионов
1-я группа катионов
В первую аналитич.гр.катионоввход.ионов калия K+, натрия Na+, аммония NH4+ и магния Mg2+. В отличии от катионов др.групп боль-во солей калия, натрия, аммония, легко растворимы в воде. Ион Mg2+ св-вами несколько отлич.от других катионов этой группы. Он образ.труднорастворимые в воде гидрат окиси, фосфорнокислую и углекислую соли. Поскольку нерастворимость в воде углекислых солей – важнейший аналитич.признак катионов 2-й группы, то Mg2+ иногда относят к ней.
Реакции катионов калия
Реакция с кобальтинитритом натрия Na3[Co(NO2)6].
Кобальтинитрит натрия в нейтр.или уксусном р-ре дает с ионами калия желтый кристаллич. осадок кобальтинитрита калия-натрия:
2KCl + Na3[Co(NO2)6] = K2Na[Co(NO2)6] + 2NaCl
иливионномвиде:
2K+ +Na+ + [Co(NO2)6]3- = K2Na[Co(NO2)6]
Реакции катионов аммония
Реакция с реактивом Несслера
(щелочной раствор ртутноиодистого калия K2[HgI4]).
Этот реактив дает с аммонийными солями красновато-коричневый осадок состава [NH2Hg2O]I (его структурная формула HO – Hg –NH – I ):
NH4Cl + 2 K2[HgI4] + 4KOH = [NH2Hg2O]I + 7KI + KCl + 3H2O
или в ионном виде:
NH4+ + 2[HgI4]- + 4OH- = [NH2Hg2O]I + 7I- + 3H2O
При очень малых колич солей аммония вместо осадка получ.желтый раствор. Реакция очень чувствит.
2-ая группа катионов
Ко 2-ой аналитич.группе катионов относятся ионы Ba2+, Ca2+, Sr2+.
Их называют щелочноземельными металлами. По своей активности немного уступают щелочным металлам. Щелочноземельные металлы образ.большоеколич.солей; из них растворимы галоидные, азотнокислые, уксуснокислые и кислые углекислые. Групповой реактив– углекислый аммоний (NH4)2CO3, образ.с ионами Ba2+ и Ca2+ не растворимые в воде средние соли BaCO3 и CaCO3.
Реакции катионов кальция
Реакция с ферроцианидом калия K4[Fe(CN)6].
Этот реактив с солями кальция в присутствии солей аммония образ. белый кристаллический осадок ферроцианида кальция и аммония Ca(NH4)2[Fe(CN)6]:
CaCl2 + 2NH4Cl + K4[Fe(CN)6] = Ca(NH4)2[Fe(CN)6] + 4KCl
или в ионном виде:
Ca2+ + 2 NH4+ + [Fe(CN)6]4- = Ca(NH4)2[Fe(CN)6]
3-я группа катионов
К 3-ей аналитич.группе катионов относ.ионы Al3+, Cr3+, Fe2+, Fe3+, Mn2+, Zn2+.
Сернистые соед.катионов этой группы не растворимы в воде, но растворимы в разбавленных в минеральных. Вследствие этого сероводород не осаждает катионы 3-ей группы из кислых р-ров. Поэт.для полного осаждения катионов 3-ей группы в виде сернистых соединений вместо сероводорода прим. его хорошо диссоциированные соли. Групповой реактив – сернистый аммоний (NH4)S. Хлористые, сернокислые, и азотнокислые соли этих элементов растворимы в воде. Растворы их вследствие гидролиза имеют слабокислую реакцию.
Реакции катионов трехвалентного железа
Реакция с гексацианоферратом (II) калия K4[Fe(CN)6]3.
K4[Fe(CN)6] даёт с солями Fe3+ в кислой среде синий осадок называемой берлинской лазури:
4FeCl3 + 3 K4[Fe(CN)6] = Fe4[Fe(CN)6]3 + 12KCl
или в ионном виде:
4Fe3+ + 3[Fe(CN)6] = Fe4[Fe(CN)6]3
Реакции катионов двухвалентного железа
Реакция с гексацианоферратом (III) калия K3[Fe(CN)6].
K3[Fe(CN)6], назыв.красной кровяной солью, дает с солями Fe2+ в кислой среде темно-синий осадок железосинеродистой закиси железа (турнбулева синь) Fe3[Fe(CN)6]2:
3FeSO4 + K3[Fe(CN)6] = Fe3[Fe(CN)6]2 + K2SO4
или в ионном виде:
3Fe2+ + [Fe(CN)6]3- = Fe3[Fe(CN)6]2
Реакции катионов цинка
Реакция с гексацианоферратом (II) калия K4[Fe(CN)6]3.
K4[Fe(CN)6] образует с ионами цинка белый осадок железистосинеродистого калия и цинка:
3ZnCl2 + 2K4[Fe(CN)6] = Zn3K2[Fe(CN)6]2 + 6KCl
или в ионном виде:
3Zn2+ + 2 K+ 2[Fe(CN)6] = Zn3K2[Fe(CN)6]2
4-ая группа катионов
Относятся катионы Hg2+, Cu2+, Bi3+, Ag+, Pb2+.
Сернистые соед.этих металлов не р-римы в разбавленных кислотах. Груп.реактив-сероводород. Многие катионы 4-й группы склоны к образ.прочных комплексов с аммиаком, цианистыми соед. и другими в-вами, что с успехом использ. в аналитич. хим.
Реакции катионов меди
Реакция с гексацианоферратом (II) калия K4[Fe(CN)6]3.
K4[Fe(CN)6] выдел. из р-ра солей двухвалентной меди красно-бурой осадок железистосинеродистой меди Cu2[Fe(CN)6]:
2CuSO4 + K4[Fe(CN)6] = Cu2[Fe(CN)6] + 2K2SO4
или в ионном виде:
2Cu2+ + [Fe(CN)6]4- = Cu2[Fe(CN)6]
Осадок не ра-рим в разбавленных кислотах, но растворяется в NH4OH, образуя аммиакат меди:
Cu2[Fe(CN)6] + 12NH4OH = 2[Cu(NH4)3](OH)2 + (NH4)4[Fe(CN)6] + 8H2O
или в ионном виде:
Cu2[Fe(CN)6] + 8NH3 = 2[Cu(NH4)3]2+ + [Fe(CN)6]4-
5-ая группа катионов
К 5-ой аналитич.группеотнос.катионы мышьяка, сурьмы, олова.
Групп.реактив–многосернистый аммоний. Многосернистый аммоний готовят, растворяя серу в сернистом аммонии. Он является окислителем.Люб.комплекс.соед.сост. из центр.атома и координированных вокруг него частиц, кот.назыв.лигандами. Хим.связь между центр.атомом и лигандом носит донорно-акцепторный характер, причем донором пары электронов явл.лиганд, а акцептором–центр.атом. Лиганд может иметь несколько донорных атомов, способных образ.хими.связь с центральным атомом. По этому признаку они делятся на монодентатные и полидентатные. Монодентатныйлигандзаним. одно координационное место у центрального атома; полидентатный – несколько: два, три и т.д. Макс. число монодентатныхлигандов, кот.м. разместиться вокруг центр.атома, носит название координационного числа атома комплексообразователя. Центр.атом и располож.вокруг него лиганды образ. внутр.координационную сферу, кот.иногданазыв.первой координационной сферой. Внутр.координационная сфера м.иметь положит., отрицат. или нулевой электрический заряд. Если внутр.координационная сфера имеет заряд, мы имеем дело с комплексным катионом или анионом, и для электронейтральностикомпл.соед.должносодерж.анионы или катионы, которые размещ.во внешней или второй координационной сфере. Связь между внутр. и внешней координационными сферами носит чисто ионный характер. Поэт.в водных р-рах ионы, наход.во внешней координационной сфере комплекса, полностью диссоциированы.Лигандыпредст.собой анионы или полярные молекулы. К неорг.лигандам относ. молекулы воды и аммиака, а также гидроксид-, галогенид-, цианид-ионы и т.д. Одним из наиболее распространенных лигандов является аммиак. Комплексы с орг.лигандами интенсивно окрашены, нер-римы в воде и легко р-римы в органич. средах. Обычно лиганды содержат такие донорные атомы, как кислород, азот, сера, фосфор и мышьяк, вход.в состав функцион.группорганич.реагентов.
В комплексах с полидентатнымилигандами могут образ.хелатные циклы. Такие комплексы называют хелатами. Хелаты, в кот.замыкание цикла происходит в результате вытеснения ионом металла одного или нескольких протонов из кислотных групп лиганда, называют внутрикомплексными соед.
11. Гидролиз солей Гидролиз солей - взаимодействие ионов соли с ионами воды, приводящее к образованию слабого электролита. степень гидролиза-отношение части соли, подвергающейся гидролизу, к общей концентрации её ионов в растворе ?=(cгидр/cобщ)·100 %где cгидр — число молейгидролизованной соли, cобщ — общее число молей растворённой соли.Степень гидролиза соли тем выше, чем слабее кислота или основание, её образующие.
Константа гидролиза — константа равновесия гидролитической реакции. Выведем уравнение константы гидролиза соли, образованной слабой кислотой и сильнымоснованием 
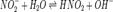 Уравнение константы равновесия для данной реакции будет иметь вид:
Уравнение константы равновесия для данной реакции будет иметь вид: 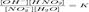 или
или 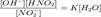 Различают:1. Гидролиз соли слабой кислоты и сильного основания:Na2CO3 + Н2О = NaHCO3 + NaOHCO32? + H2O = HCO3? + OH?( щелочная среда, реакция обратима)2. Гидролиз соли сильной кислоты и слабого основания:CuCl2 + Н2О = CuOHCl + HCl Cu2+ + Н2О = CuOH+ + Н+(кислая среда, реакция протекает обратимо) 3. Гидролиз соли слабой кислоты и слабого основания:Al2S3 + 6H2O = 2Al(OH)3 + 3H2S 2Al3+ + 3S2? + 6Н2О = 2Al(OH)3(осадок) +ЗН2S(газ) Соль сильной кислоты и сильного основания не подвергается гидролизу, и нейтрален
Различают:1. Гидролиз соли слабой кислоты и сильного основания:Na2CO3 + Н2О = NaHCO3 + NaOHCO32? + H2O = HCO3? + OH?( щелочная среда, реакция обратима)2. Гидролиз соли сильной кислоты и слабого основания:CuCl2 + Н2О = CuOHCl + HCl Cu2+ + Н2О = CuOH+ + Н+(кислая среда, реакция протекает обратимо) 3. Гидролиз соли слабой кислоты и слабого основания:Al2S3 + 6H2O = 2Al(OH)3 + 3H2S 2Al3+ + 3S2? + 6Н2О = 2Al(OH)3(осадок) +ЗН2S(газ) Соль сильной кислоты и сильного основания не подвергается гидролизу, и нейтрален
Дата добавления: 2019-09-02; просмотров: 565; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!