Гены и наследственные болезни 14 страница
Впоследствии выяснилось, что своих предшественников имеют не только кровяные клетки. Эпителий кишечника, клетки кожи, мышц и сосудов, наконец костная ткань тоже все время обновляются. А чуть позже среди обычных кроветворных стволовых клеток была обнаружена популяция еще более пластичных, умеющих трансформироваться абсолютно в любую клетку человеческого организма (их у нас насчитывается около 290 типов). Короче говоря, такая плюрипотентная[47] стволовая клетка может дать начало любой ткани нашего тела. Медики возлагают на них большие надежды.
Сравнительно недавно стволовые клетки особого типа были найдены в раковых опухолях. Оказалось, что свыше 90 % опухолевой ткани приходится на обычные «старые» раковые клетки, неспособные к развитию, а ее бурный рост обусловлен размножением стволовых опухолевых клеток. Удельный вес этих клеток при разных формах рака сильно варьируется. Например, в случае лейкемии[48] на 100 тысяч злокачественных клеток приходится всего лишь одна стволовая, а при некоторых мозговых опухолях их число может достигать 20. Понятно, что агрессивность опухоли напрямую связана с числом стволовых клеток: чем их больше, тем быстрее опухолевый рост.
По мнению ученых, это самые обычные стволовые клетки, переродившиеся в результате мутации и ставшие смертельно опасными, ибо в полной мере унаследовали весьма эффективный механизм репарации ДНК. Поэтому традиционные методы лечения – радио-или химиотерапия – сплошь и рядом оказываются бессильны: проходит немного времени, и опухоль воскресает, как феникс из пепла.
А совсем недавно была высказана гипотеза, согласно которой причина рака кроется в пробуждении «спящих» генов клеточной кооперации. Эти гены появились около миллиарда лет назад одновременно с возникновением многоклеточных организмов.
Вероятно, читателю приходилось слышать об атавизмах – внезапном проявлении признаков, свойственных нашим далеким предкам. Объясняется это тем, что гены, за них отвечающие, продолжают сохраняться в геноме, но либо отключены, либо представляют собой некодирующие фрагменты ДНК. Эволюционно более молодые гены зорко приглядывают за дремлющими «старичками», а когда в силу тех или иных причин теряют бдительность, на свет божий появляются хвостатые и волосатые младенцы или детишки с перепонками между пальцами. Зубы у цыплят тоже воз никают в результате несвоевременного пробуждения древних генов. Одним словом, авторы новой гипотезы полагают, что рак – это своеобразный атавизм, активизация спящих генов, прекративших работу более 600 миллионов лет назад.
Переход от одноклеточных организмов к многоклеточным свершился не в одночасье, а растянулся на десятки миллионов лет. Дифференцировки органов и клеточной специализации тогда не существовало, так что первые организмы, ступившие на коварную тропу многоклеточности, представляли собой простонапросто рыхлые колонии эукариот, клеточное взаимодействие которых ограничивалось обменом химическими сигналами. Одним словом, эти бурно растущие конгломераты весьма напоминали раковую опухоль. А вот когда в кембрии[49] (или венде) появились сложные организмы, возникла необходимость координировать деятельность большого количества клеток разных типов. Однако природа – слепой конструктор, действующий методом проб и ошибок, – ничего не создает с нуля, она только лишь видоизменяет сделанное ранее. Поэтому новые многоклеточные унаследовали геном своих одноклеточных предков. Конечно, эволюция его основательно пошлифовала: одни гены были откорректированы, а другие вообще умолкли, но древние механизмы существуют в геноме до сих пор. И если в организме происходит сбой, доисторические гены могут активироваться и вызвать злокачественный рост. Так что опухоль – это древнее многоклеточное образование, которое может поселиться в каждом из нас.
Впрочем, у авторов красивой гипотезы впереди много работы. Для начала неплохо бы разобраться с некоторыми из ныне живущих, но очень древних организмов, которые умеют восстанавливаться из мельчайших фрагментов. Например, обыкновенную пресноводную гидру (примитивное животное из группы кишечнополостных) можно потереть на терке, и она легко вырастет вновь, чуть ли не из отдельной клетки. Точно так же обстоит дело и с губками. Кстати, расшифровка генома одной из губок выявила участки ДНК, отвечающие за рудиментарную клеточную кооперацию и за регуляцию роста. Неполадки в работе этих генов у высших организмов как раз и вызывают бесконтрольное размножение опухолевых клеток.
Одним словом, какая бы версия развития рака ни восторжествовала, бесспорно одно: наследственная предрасположенность играет здесь немалую роль.
И похоже, что окончательно победить этот недуг можно только путем генного модифицирования.
Некоторые психические болезни (шизофрения, в частности) тоже имеют наследственную природу – сегодня уже выявлены десятки генов, мутации в которых повышают риск развития тяжелого заболевания.
Однако не все так безнадежно и неопределенно. На сегодняшний день известно много болезней, тип наследования которых хорошо изучен. Вот, скажем, гемофилия – врожденный дефект свертывания крови, опасный недуг, которым страдал царевич Алексей, сын Николая II. В развитии этой болезни ключевую роль играют специфические белки – так называемые факторы свертываемости, и если в двух генах, их кодирующих, произошла мутация, нормальных белков в организме просто нет.
Оба гена располагаются в X-хромосоме, так что это наследование, сцепленное с полом. Поскольку у женщин две X-хромосомы, присутствие мутантной версии гена гемофилии в одной хромосоме компенсируется нормальным геном в другой. Поэтому женщины гемофилией практически не болеют, так как вероятность того, что в обеих хромосомах окажется «порченый» ген, исчезающе мала. А вот мужчинам скомпенсировать дефектный ген нечем – в Y-хромосоме его просто-напросто нет. Поэтому гемофилия – сугубо мужская болезнь. Кроме царевича Алексея этим недугом страдали испанские принцы и родные братья прусского принца Сигизмунда. От гемофилии умер брат английского короля Эдуарда VII. Одним словом, «королевская» болезнь в чистом виде – едва ли не все европейские дворы могли «похвастаться» собственными гемофиликами.
Причина между тем проста. Династические традиции (весьма и весьма жесткие) обязывали несчастных принцев жениться только на принцессах сопредельных дворов. А поскольку принцесс на свете не так много, то все царствующие фамилии Европы находились в кровном родстве. Судя по всему, носительницей гена гемофилии была английская королева Виктория, просидевшая на троне без малого 70 лет – с 1837 года по 1901-й. А так как никто из ее предков и родни по боковым линиям гемофилией не болел, резонно предположить, что генная мутация в X-хромосоме произошла у одного из ее родителей или у нее самой на ранней стадии эмбрионального развития.
У Виктории было девять детей. От гемофилии в возрасте 31 года умер один из ее сыновей – Леопольд, брат Эдуарда VII, о котором мы упомянули чуть выше. С принца Альберта – мужа королевы – подозрения снимаются, ибо он был здоров, а передача гена гемофилии от отца к сыну невозможна. Эта хворь наследуется от дяди к племяннику, что говорит о рецессивной мутации в X-хромосоме. От британской королевы-долгожительницы ген гемофилии унаследовали две ее дочери, поэтому многие их сыновья оказались больны. Алиса Гессенская, одна из дочерей Виктории, была носительницей опасного гена и матерью Александры Федоровны – жены последнего российского императора Николая II. Царевич Алексей унаследовал ген гемофилии именно от нее.
Между прочим, гемофилия – заболевание, известное с древнейших времен. Сообщения о ней в Талмуде относятся к VI веку до н. э. Уже тогда предписывалось воздерживаться от обрезания мальчиков, у старших братьев которых после этой процедуры наблюдалось сильное кровотечение. Сыновей сестры той женщины, чей сын терял много крови, тоже не обрезали. А вот если у того же отца были сыновья, рожденные от другой женщины, то на них запрет не распространялся. Совершенно очевидно, что тип наследования гемофилии был в общих чертах ясен задолго до рождества Христова. Так что поведение европейских королей (и российского царя, в частности) следует квалифицировать как преступную халатность или глупый аристократический гонор.
В своей книжке «Почему я похож на папу» Н. В. Лучник приводит еще более впечатляющий пример стойкости наследственных признаков в ряду поколений. Полководец английской армии Джон Тальбот, павший на поле брани в 1453 году, на излете Столетней войны, был с почестями похоронен в фамильном склепе Шрюберийского собора (король пожаловал ему незадолго до этого титул герцога Шрюберийского). А в 1914 году затеяли реставрацию собора, причем ремонтные работы возглавлял прямой потомок Тальбота, тринадцатый герцог Шрюберийский. У него была синфалангия – срастание первой и второй костной фаланги на пальцах руки, врожденный дефект, унаследованный от отца. Каково же было его удивление, когда он увидел на руке полуистлевших останков далекого предка в точности такой же изъян – сросшиеся фаланги пальцев. Родовой признак благополучно пережил 14 поколений.
Современная медицина насчитывает чуть более 5000 наследственных заболеваний. Лучше всего изучены хромосомные аномалии, когда меняется число хромосом или в них самих обнаруживаются нарушения – выпадение одного участка (делеция), его поворот на 180 градусов (инверсия) или его перемещение в другую хромосому (транслокация).
Например, трисомия по 21-й паре хромосом (то есть к 21-й паре присоединяется маленький «довесок» в виде дополнительной хромосомы) ведет к развитию синдрома Дауна. Такие дети сразу обращают на себя внимание малым ростом, короткопалостью, своеобразной «дальневосточной» раскосостью, монгольской складкой века (эпикантусом) и вообще монголоидной уплощенностью черт лица. Кроме того, у них отмечаются пониженный тонус мышц, недостаточность желез внутренней секреции и выраженная умственная отсталость.
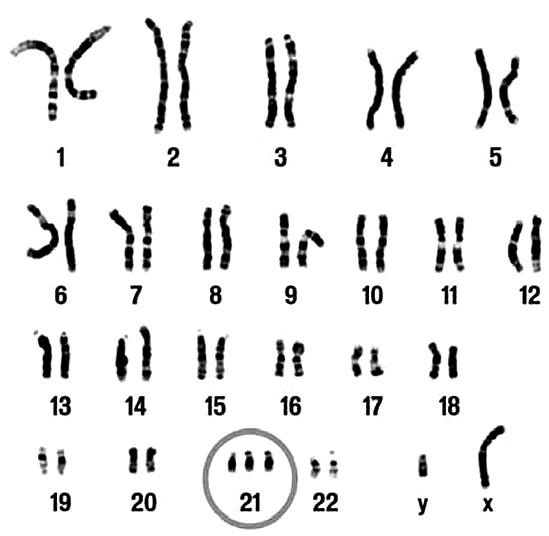
Трисомия по 21-й паре хромосом
А трисомия по 13-й паре приводит к рождению детей с резким недоразвитием головного мозга и тяжелейшими пороками сердца, которые умирают в младенческом возрасте. Лишняя половая хромосома вызывает синдром Клайнфельтера, проявляющийся задержкой полового развития, умственной отсталостью и затрудненной речью. Чаще всего у больных с этим синдромом обнаруживают классический набор – XXY, но экзотические наборы XXYY, XXXY и даже XXXXY дают ту же самую клиническую картину.
Если диагностика хромосомных аномалий особых трудностей сегодня не представляет, то с точковыми мутациями, затрагивающими отдельные гены, дело обстоит значительно хуже. Кроме того, многие болезни, имеющие наследственную природу, обусловлены «неправильной» работой нескольких десятков генов (некоторые психические болезни, например шизофрения). Тем не менее, уровень развития современных биотехнологий позволяет хотя бы в принципе нащупать терапевтические подходы к лечению врожденных недугов, а порой даже получить весьма неплохие результаты.
Генная терапия некоторых наследственных болезней – задача, вполне решаемая уже сегодня. Несколько лет назад появились сообщения об успешных генно-инженерных вмешательствах при муковисцидозе и тяжелых комбинированных иммунодефицитах (ТКИД). Муковисцидоз – неизлечимая наследственная болезнь, при которой происходит кистозное перерождение поджелудочной железы, желез кишечника и дыхательных путей в результате закупорки их выводных протоков вязким слизистым секретом. А ТКИД развивается из-за дефекта синтеза особого фермента, что приводит к накоплению токсических продуктов, убивающих T– и B-лимфоциты – центральное звено системы иммунитета. Для таких больных смертельно опасна любая, самая безобидная инфекция, и они вынуждены проводить всю свою жизнь в изолированной от внешнего мира стерильной камере. Но сначала несколько слов о методах генной инженерии.
Полезный ген вырезают из хромосомы одного организма и встраивают его в хромосому другого, в любую из его клеток. Казалось бы, нехитрая операция, но она проста только на первый взгляд. Сначала «донорский» ген нужно отыскать и вырезать его из молекулы ДНК с помощью специальных ферментов-рестриктаз. А для доставки гена по адресу – внутрь чужой клетки – используют так называемые векторы – «отредактированные» фаги, вирусы и плазмиды (кольцевые молекулы ДНК). С натуральными вирусами работать нельзя: они могут проявить свои агрессивные качества, и тогда все пойдет насмарку. Поэтому сначала их подвергают своеобразной хирургической операции, вырезая все лишнее и оставляя только те гены, которые доставят груз по назначению.
Итак, теперь в распоряжении биологов есть полезный ген и средство доставки. Остается вложить письмо в конверт. Для этого кольцевую молекулу ДНК (плазмиду) режут в нужном месте рестриктазами, внедряют в нее копию гена и запечатывают конверт – возвращают плазмиде исходную форму с помощью сшивающих ферментов – лигаз.
Когда полезный ген окажется в нужном месте чужой клетки, нужно убедиться, что он «прижился» и успешно работает (детали этой процедуры мы опустим). Если объектом был микроорганизм, то задача выполнена: создана популяция трансгенных клеток, готовых синтезировать несвойственный им продукт. Таким образом уже давно получают инсулин из бактерий и дрожжевых клеток, в геном которых вставлен соответствующий человеческий ген. Сложнее с растениями, потому что из культуры клеток их нужно сначала вырастить, и еще сложнее с животными, так как приходится работать с оплодотворенной яйцеклеткой, а потом еще вдобавок подыскивать для нее суррогатную мать.
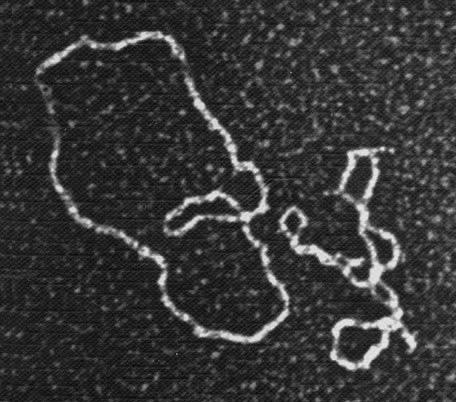
Плазмиды, визуализированные с помощью электронного микроскопа
В целях генной терапии муковисцидоза для корректировки дефектного гена использовали векторхимеру – тщательно обезвреженный вирус СПИДа (чтобы не развилась инфекция) и белковую оболочку вируса лихорадки Эбола (смертельно опасная тропическая инфекция), потому что вирус иммунодефицита не умеет самостоятельно размножаться в клетках легочного эпителия.
Биолог Александр Чубенко пишет:
Испытания на мышах и обезьянах показали высокую эффективность гибридного вируса: правильный ген удалось внедрить почти в четверть клеток легочного эпителия. Однако излечиться раз и навсегда таким способом невозможно. Вирус исправляет геном только в поверхностных клетках легочного эпителия, и лечение необходимо повторять несколько раз в год по мере отмирания клеток.
А вот при лечении ТКИД в качестве вектора использовался ретровирус, и у двоих из 12 детей развилась лейкемия. Но большинство специалистов всетаки решили, что в данном случае риск оправдан, так как без лечения смерть гарантирована, а лейкоз – меньшее зло. Для доставки генов в клетки-мишени применяются и другие модифицированные вирусы (вирус герпеса, аденовирусы), и хотя результаты, бесспорно, впечатляют, иногда пробежит холодок по спине: кто знает, какие побочные эффекты может вызвать этот вирус…
Правда, существуют и альтернативные, невирусные способы переноса генетического материала в клетку (например, с помощью липосом, умеющих просачиваться через клеточную мембрану, специфических антител и путем непосредственной бомбардировки клеток микрочастицами золота, к которым присоединены фрагменты ДНК), но все эти методы гораздо менее надежны. В клеточные ядра попадает ничтожная часть терапевтических генов, а в хромосомы они встраиваются с большим трудом.
В последнее время активно разрабатываются генные технологии по созданию искусственных хромосом с последующим их внедрением в клетки-мишени. Эксперименты на животных дают неплохие результаты, и ученые считают, что конструирование искусственных хромосом со встроенным лечебным геном для терапии наследственных болезней человека – дело ближайшего будущего. Вот что пишут специалисты:
Вводить хромосомы в ядро можно будет либо заключив их в контейнеры-липосомы, либо с помощью инъекций иглой атомно-силового микроскопа. Японские нанотехнологи в ноябрьском номере журнала Nano Letters за 2004 год опубликовали статью, в которой описан зонд длиной 8 микрометров и шириной 200 нанометров. В оболочке ядра клетки после прокола такой иглой образуется брешь диаметром 1 микрометр, которая исчезает после извлечения иглы. Таким способом можно проводить нанохирургические операции с генетическим материалом непосредственно в живых клетках без нарушения целостности их микроструктур.
Бум генной терапии пришелся на середину 1990-х годов, когда «отремонтированные» кроветворные клетки удалось пересадить детям с тяжелой формой врожденного иммунодефицита. На первых порах все шло хорошо (детей спасли от неминуемой смерти), но у двух больных развилась лейкемия. Откуда взялась эта напасть?
Американские биологи создали дрожжевую культуру, клетки которой содержат как обычные, так и синтетические хромосомы
Беда в том, что доставить нужный ген по адресу – всего лишь полдела. Нужно еще научить клетку читать вирусный белок. С другой стороны, вирус («отредактированный» вектор) тоже хочет быть прочитанным. Для этой цели у него есть промотор – особый фрагмент ДНК, который, взмахнув флажком, дает команду: поехали! Но вирусный промотор не подчиняется клеточным регуляторам, поэтому клетка читает не только полезный ген, но и соседние, оказавшиеся рядом. А среди них легко может оказаться латентный (скрытый) онкоген, запускающий злокачественное перерождение клетки. А поскольку каждому ребенку вводят около миллиона таких химер, вероятность того, что плохой ген вдруг неожиданно «выстрелит», резко увеличивается. Поэтому многообещающую программу без лишнего шума свернули.
Однако совсем недавно французские и американские ученые объявили, что разработана надежная генно-инженерная методика лечения талассемии[50] – врожденного заболевания крови. Тяжелые формы талассемии заканчиваются смертью больных в раннем детстве. Выбор у них невелик: или регулярные гемотрансфузии (переливания крови), или пересадка донорского костного мозга. В первом случае больной обречен на пожизненные процедуры, а во втором его подстерегает хронический иммунный конфликт. Но ученые сумели отыскать третий, неочевидный, путь. Они внедрили в клетки костного мозга больного неповрежденную версию гена, после чего модифицированные кроветворные клетки были возвращены на «родину» и занялись своим прямым делом – созреванием и размножением. Результат превзошел все ожидания:
Через год после лечения пациент смог отказаться от переливаний крови и вот уже 21 месяц успешно обходится без них, хотя прежде вынужден был делать их ежемесячно, начиная с трехлетнего возраста (больному на момент операции исполнилось 15 лет. – Л.Ш .). По сути дела, речь идет о радикальном и полном излечении от тяжелой генетической болезни.
Но бить в литавры пока рано. Авторы, к сожалению, не пишут о том, каким образом им удалось выявить и нейтрализовать латентные онкогены. Быть может, притаившаяся лейкемия еще покажет себя во всей красе. Или не покажет, так как выборка минимальна – один-единственный человек. А вот когда счет пойдет на десятки, тогда и поговорим.
Дата добавления: 2019-02-13; просмотров: 315; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!