Комбинированные иммунодефициты
Синдром Вискотта-Олдрича (WASотWiskott-Aldrich syndrome)– Х-сцепленный синдром, который наследуется по рецессивному типу. Обусловлен дефектом гена в участке Хp11.22-11.23, кодирующего белок WASP (Wiskott-Aldrich syndrome protein), который участвует в полимеризации актина и формировании цитоскелета, действует как платформа для сигнальных молекул. Изменяется экспрессия CD43 (уменьшается на Т-лимфоцитах). Отсутствие белка WASP в лимфоцитах приводит к нарушению функций Т-клеток и регуляции синтеза антител.
Клиника: этот Х-связанный дефект встречается у мальчиков. Характерна триада признаков: экзема кожи, тромбоцитопеническая пурпура и высокая восприимчивость к инфекциям. Другие проявления: тяжелый аутоиммунный васкулит или гломерулонефрит и высокая частота злокачественных заболеваний, вовлекающих лимфоциты, например, лимфомы и лейкемии.
Лабораторные критерии: подсчет тромбоцитов обнаруживает тромбоцитопению с маленькими тромбоцитами и аномальной экспрессией поверхностных гликопротеинов CD43 и GPIb. Уровень IgG в плазме обычно нормален, IgA и IgЕ часто повышены, снижены IgM, особенно у старших детей. Пациенты с WAS имеют низкиеуровни антител к антигенам группы крови и не в состоянии продуцировать антитела против некоторых вакцин, содержащих полисахариды или сложные сахара. Кожные тесты на функцию Т-лимфоцитов могут дать отрицательный ответ, функция Т-лимфоцитов может быть аномальной, снижен пролиферативный ответ на митогенную стимуляцию. Количество Т-лимфоцитов прогрессивно снижается. Количество В-лимфоцитов в норме.
|
|
|
Атаксия-телеангиоэктазия (Синдром Луи-Бар) – аутосомно-рецессивный тип наследования. Мутация гена в хромосоме 11 (11q22-23), продукт которого – белок семейства внутриядерных ДНК-зависимых протеинкиназ, имеющий отношение к контролю клеточного цикла, трансдукци митогенных сигналов и рекомбинациям при мейозе. Клетки высокочувствительны к ионизирующей радиации, в них нарушена репарация ДНК и повышены хромосомные аномалии. Отмечается гипоплазия тимуса, селезенки, лимфоузлов.
Клиника: Развивается мозжечковая атаксия, телеангиэктазия склеры, конъюнктивы и кожи, хронические инфекции околоносовых пазух и легких, злокачественные опухоли. Заболевание чаще диагностируется в возрасте 5 – 7 лет.
Лабораторные критерии:
Снижен уровень иммуноглобулинов (IgA, IgЕ и субклассы IgG (IgG2 и IgG4), IgM повышены, наблюдается вариабельное снижение АТ. Количество В-лимфоцитов в норме. Количество и функциональная активность CD4+ Т-лимфоцитов снижены. Отрицательный тест на ГЗТ. Уровень α-фетопротеина повышен.
Тяжелые комбинированные иммунодефициты – ТКИД(severe combined immunodeficiencies – SCID) составляют около40% всех первичных иммунодефицитов. Связаны смножественными дефектами генов. Различают Х-сцепленный ТКИД и ТКИД аутосомно-рецессивным типом наследования.
|
|
|
Молекулярные дефекты при ТКИД и особенности фенотипа лимфоцитов (по Rebecca H. Buckley, 2003)
| Молекулярный дефект | Фенотип лимфоцитов |
| Х-сцепленный ТКИД | |
| 1. Дефекты гена γ-цепи | Т(-)В(+)NK(-) |
| ТКИД с аутосомно-рецессивным типом наследования | |
| 1.Мутация гена АДА | Т(-)В(-)NK(-) |
| 2. Мутация гена JAK3 | Т(-)В(+)NK(-) |
| 3. Мутация гена IL-7Rα-цепи | Т(-)В(+)NK(+) |
| 4. Мутация RAG-1 и RAG-2 | Т(-)В(-)NK(+) |
| 5. Мутация гена Artemis | Т(-)В(-)NK(+) |
| 6. Мутация гена CD45 | Т(-)В(+) |
Х-сцепленный ТКИД – нарушения, связанные с мутацией гена, локализованного в хромосоме Х (Xq13). Данный ген кодирует структуру
общей гамма-цепи (γc) рецепторов для цитокинов (ИЛ-2, -4, -7, -9, -15, -21). на поверхности Т- и NK-лимфоцитов. В результате мутации гена нарушается передача сигналов при связывании рецепторов с цитокинами, созревание и функционирование Т-лимфоцитов..
Клиника: Болеют мальчики, проявляется в первые недели жизни ребенка неуправляемыми поносами, повторными инфекциями, задержкой нарастания массы тела (гипотрофия II-III ст). Гибель (как правило) в первые 2 года жизни. Полагают, что 10% детей, погибающих на первом году жизни, страдали ТКИД.
|
|
|
ТКИД часто называется «болезнью пузырного мальчика». Это заболевание стало широко известным в течение 1970-80-ых годов, когда мир узнал о Дэвиде Веттере, мальчике с Х-связанным SCID, который жил в течение 12 лет в пластмассовом стерильном контейнере.
Лабораторные критерии: Уровень IgА, IgМ, IgG значительно снижен.Типичны очень низкие количества Т- и NK-лимфоцитов, при нормальном количестве В-клеток. Функции Т- и В-лимфоцитов снижены.
Лечение: Трансплантация костного мозга, совместимого по HLA. Разрабатывается генотерапия путем введения гена γ-цепи в костномозговые клетки.
Недостаточность аденозиндезаминазы (синдром АДА).Ген АДА находится в хромосоме 20q13-ter. Дефицит АДА способствует накоплению токсических метаболитов пути пурина (dATP) и пути метилирования (S-аденозилгомоцистеин), которые накапливаются в клетке и вызывают апоптоз тимоцитов и циркулирующих лимфоцитов.
Клиника: В первые недели жизни – лимфоцитопения, анемия. Характерны множественные дефекты костей скелета – хондроостеодисплазия, выявляемая с помощью радиографического исследования. Отмечается низкое содержание АДА в эритроцитах и лимфоцитах.Инволюция вилочковой железы.
|
|
|
Лабораторные критерии: Т-, В-лимфоциты и Ig-ны не обнаруживаются.
Лечение: Трансплантация костного мозга, полностью совместимого по HLA. Перед пересадкой из донорского костного мозга удаляют Т-лимфоциты. Эффективна ферментозаместительная терапия, проводимая подкожным введением АДА крупного рогатого скота, конъюгированной с полиэтиленгликолем (нецелесообразно начинать введение препарата при предполагаемой трансплантации костного мозга). Возможна генотерапия путем переноса гена АДА в культивируемые Т-лимфоциты больного. Размеры гена АДА средние, он хорошо встраивается в ретровирусные векторы, вводимые в активированные лимфоциты больных.
Дефицит Янус-киназы (JAK3).У больных выявляется мутация гена, кодирующего структуру JAK3 – единственной сигнальной молекулы, передающей сигнал для γ-цепи, сопряженной со многими рецепторами цитокинов. Иммунный статус таких больных близок к показателям, определяемым при Х-сцепленном ТКИД: количество В-лимфоцитов повышено, а Т- и NK-клеток – понижено.
Дефицит системы фагоцитов
Синдром Чедиака- Хигаси -редкое заболевание с аутосомно-рецессивным типом наследования, при котором нарушается хемотаксис и отсутствует внутриклеточный лизис бактерий.
• В нейтрофилах, моноцитах и некоторых других клетках появляются аномально крупные везикулы, лизосомы. Гигантские лизосомы способны к слиянию с фагосомами, но не могут освобождать активные лизосомальные ферменты, поэтому нарушается процесс разрушения бактерий, фагоцитоз становится неэффективным. Нарушение хемотаксиса обусловлено дисфункцией микротрубочек цитоскелета.
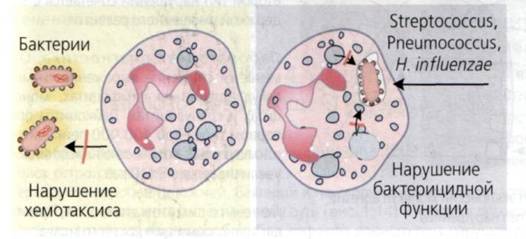
Рис.8. Синдром Чедиака-Хигаси
Клиника: Развиваются тромбоцитопения и нейтропения, лихорадка, высокая предрасположенность к инфекциям (особенно вызванным каталазанегативными бактериями), неврологические нарушения, светобоязнь, а также частичный альбинизм кожи и волос (изменения в меланоцитах), поэтому волосы и кожа пациента серебристо-светлые). Повышается кровоточивость (изменения в тромбоцитах).
Лабораторная диагностика: Выявляют нарушения хемотаксиса и фагоцитоза нейтрофилов. В нейтрофилах, окрашенных на миелопероксидазу, выявляются гигантские гранулы. Уменьшается количество NK-клеток.
Лечение:симптоматическое.
Хроническая гранулематозная болезнь (ХГБ) –развивается у детей с наследственными дефектами бактерицидной активности фагоцитов (макрофагов и нейтрофилов): утрачивается способность клеток вырабатывать антимикробные метаболиты кислорода и уничтожать каталазаположительные микробы.
Фагоцитированные микробы не уничтожаются, в результате чего развиваются повторные гнойные инфекции кожи, подкожной клетчатки, легких, печени и и других органов и тканей. Фагоциты превращаются в «хранилища» для микробов, способствуя хронизации процесса. Скопления клеток образуют гранулемы. Дефект обусловлен недостатком цитохрома b558 в мембране фагосомы гранулоцтов. Никотинамиддинуклеотидфосфат (НАДФ) не способен транспортировать электроны, необходимые для образования радикалов кислорода и передавать их молекулам О2. Дефект локализован в хромосоме Xp21. НАДФ-оксидаза выполняет ключевую роль в этой окислительно-восстановительной реакции, катализируя превращение О2 в супероксиданион кислорода, участвующий в бактерицидности нейтрофилов. Дефект НАДФ-оксидазы и недостаток глюкозо-6-фосфатдегидрогеназы ответственны за неспособность гранулоцитов убивать фагоцитированные бактерии.
Обычно встречается Х-сцепленная форма хронического гранулематоза, характеризующаяся дефектом цепи 91 кДа в составе b558. Другие три типа являются аутосомно-рецессивными расстройствами, возникающими в результате дефектов другой цепи цитохрома b558 либо одного из двух белков(p47phox или p67 phox). В итоге нарушается способность клеток вырабатывать супероксиданион кислорода и перекись водорода. Каталазаположительные микробы (представители родов Stаphylocjccus, Serratia, Klebsiella и грибы рода Aspergillus) выживают из-за дефекта антимикробных кислородзависимых механизмов фагоцитов. Коагулаза-негативные штаммы рода Streptococcus и вида Haemophilus influenza погибают в фагосомах от небольшого количества синтезируемой перекиси водорода.
Клиника: У больных уже на первом году жизни развиваются инфекции, чаще вызываемые каталазапродуцирующими бактериями. Развиваются лимфаденит, пневмония, пиодермия, абсцессы печени, остеомиелит, персистирующая диарея, менингит, сепсис, ринит и др.
Лабораторная диагностика: Выявление дефекта образования перекисных радикалов с помощью методов хемилюминесценции и НСТ-теста нейтрофилов. При НСТ-тесте под влиянием супероксиданиона растворимый краситель нитросиний тетразолий переходит в цитоплазме клеток в нерастворимое соединение диформазан синего цвета. Его количество пропорционально активности респираторной реакции.
При ХГБ - отрицательный НСТ-тест (при атипичной форме ХГБ – от 16 до 100% слабо позитивных нейтрофилов). Отсутствует хемилюминесценция нейтрофилов.
Дата добавления: 2018-04-15; просмотров: 523; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!