Содержание домашнего задания №1
ДОМАШНИЕ ЗАДАНИЯ
ПО ФИЗИЧЕСКОЙ ХИМИИ
Федеральное агентство по образованию
Московский государственный университет
Инженерной экологии
Домашние задания
ПО ФИЗИЧЕСКОЙ ХИМИИ
Методические указания
Под редакцией проф. А.С. Лилеева
Москва
2011
Учебное издание
Составители:
Мишустин Александр Иванович,
Белоусова Кира Филипповна
Домашние задания
По физической химии
под редакцией проф. А.С.Лилеева
Подписано.в печать
Формат бум. 60 x 84 1/16.
Объем усл. п. л. . Уч.-изд. л.. Тираж 100 экз. Зак.
МГУИЭ, издательский центр,
105066, Москва, Старая Басманная ул., 21/4
УДК 541.1
ББК 24.5
М
Рецензенты:
Допущено редакционно-издательским советом МГУИЭ
Мишустин А.И., Белоусова К.Ф.
М Домашние задания по физической химии: Методические указания /Сост.: А.И. Мишустин, К.Ф.Белоусова; под ред. А.С. Лилеева. - М.: МГУИЭ, 20. — с.
Методические указания предназначены студентам, обучающимся по всем специальностям МГУЭИ, при изучении дисциплины «Физическая химия».
УДК 544
ББК 24.5
© А.И. Мишустин, К.Ф. Белоусова, 20
© МГУИЭ, 20
Домашние задания по физической химии предусмотрены программой по этой дисциплине. С соответствующими разделами теории можно ознакомиться в учебнике [1]. Назначение данных методических указаний – помочь студентам разобраться в наиболее трудных и важных вопросах теории и методах расчета, относящихся к контрольным домашним заданиям.
|
|
|
Каждое домашнее задание содержит определенный перечень вопросов. Это в основном вопросы, для ответа на которые нужно выполнить расчеты. Есть и теоретические вопросы, требующие письменного ответа. Студент выполняет свой вариант задания, выданный ему преподавателем.
При выполнении расчета нужно указать какая формула используется, какие цифры в нее подставлены, каков конечный результат, какова размерность найденной величины. При использовании сложных формул следует указывать все промежуточные цифры расчета. Графики выполняются на миллиметровой бумаге (формат А4). На осях графика должна быть указана отложенная величина, единицы ее измерения, наносятся также масштабные единицы (но не координаты точек). Масштаб следует выбирать таким, чтобы 1 см был кратен 1, 2 или 5 единицам значений или этим же значениям, умноженным на 10 ±x , где x – целое число.
Домашнее задание следует оформлять на листах формата А4. Первый лист – титульный, на нем указывается название вуза, номер и вариант домашнего задания; фамилия студента и преподавателя, год. На втором листе должны быть расположены таблицы (по предлагаемой форме) с исходными, конечными, справочными данными. Третий и последующий листы должны содержать расчеты и ответы на теоретические вопросы. Листы следует пронумеровать и скрепить скрепкой (не степлером) или поместить в файл. При исправлении ошибок, замеченных преподавателем, нужно аккуратно зачеркнуть (не замазать) неправильный ответ и написать правильный, используя, при необходимости, новый лист бумаги.
|
|
|
В расчетах, если не оговорено другое, используется система единиц СИ.
Домашнее задание № 1. «Химическая кинетика»
Элементы теории
Химические реакции, протекающие в одну стадию, называются простыми. Такие реакции различают по молекулярности
Молекулярность реакции определяется числом частиц, принимающих участие в элементарном акте химического взаимодействия.
Молекулярность может быть равна 1, 2 или 3, соответственно, различают моно- би- и тримолекулярные реакции. Для простых реакций молекулярность совпадает с порядком реакции, а порядки по компонентам совпадают с наименьшими целочисленными стехиометрическими коэффициентами в левой части химического уравнения. Фактически, если известна молекулярность реакции, то понятен и ее истинный механизм, для установления которого проводят серьезные исследования.
|
|
|
Рассмотрим кинетику простых реакций 1, 2 и 3-го порядков.
Реакции первого порядка: A ® L + M
Порядок реакции (n) равен 1. Пусть концентрация вещества А (СА) равна С, тогда скорость реакции (w=-dС/dt) в соответствии с законом действующих масс равна
-dС/dt = kС.
Латинская буква d здесь и далее обозначает знак дифференциала, то есть бесконечно малого изменения величины. Разделим переменные: -dС/С=kdt, проинтегрируем:-lnС = kt + const, определим константу интегрирования, учитывая, что в начальный момент времени: t = 0, С = С0, отсюда const = -lnС0. В итоге получаем: lnС – lnС0 =- kt,
ln (С/С0) = - kt , С = С0 e - kt . (1.1, 1.2)
Уравнения (1.1) и (1.2) называются кинетическими уравнениями реакции первого порядка в логарифмической (1.1) и экспоненциальной (1.2) форме.
Важной кинетической характеристикой процесса является время полупревращения ( t 1/2 ), то есть время, в течение которого прореагировала половина взятого вещества.
Определим эту величину для реакции первого порядка: С = 1/2С0, ln(С0/С) = ln2/kt1/2,
t 1/2 = ln 2/ k = 0,69/ k. (1.3)
|
|
|
Время полупревращения в этом случае не зависит от начальной концентрации исходного вещества.
Размерность константы скорости для реакций первого порядка: [k] = [с]-1.
Реакции второго порядка: А + В ® L.
Порядок реакции равен 2. Пусть СA = СB = С. Тогда -dС/dt = kС2, -dС/С2 = kdt. После интегрирования и с учетом начальных условий (t = 0, С = С0) получаем:
1/С – 1/С0= kt. (1.4)
Время полупревращения для реакций второго порядка обратно пропорционально начальной концентрации:
t 1/2 = 1/( k С0). (1.5)
Размерность константы скорости: [k] = [л/(моль с)].
Реакции третьего порядка: А + В + D ® L.
Величина n равна 3. Пусть СA = СB = СD = С, тогда -dС/dt = kС3, -dС/С3 = kdt. После интегрирования, учитывая начальные условия (t = 0, С = С0), получаем:
1/С2 – 1/С02= 2 kt. (1.6)
Время полупревращения для реакций третьего порядка обратно пропорционально квадрату начальной концентрации:
t 1/2 = 3/(2 k С02). (1.7)
Размерность константы скорости: [k] = [л2/(моль2 с)].
В представленных кинетических уравнениях под символом «С» понимают текущую, то есть наблюдаемую в данный момент времени молярную концентрацию соответствующих исходных веществ (С = Cт.). Существует также понятие прореагировавшей концентрации (Cпр), характеризующее ту часть концентрации данного исходного вещества, которая превратилась в конечный продукт. Прореагировавшая концентрация связана с текущей и исходной (C0) соотношением:
С0 = Cт + Cпр (1.8).
В расчетах по уравнениям (1.1-1.7) нужно знать величину константы скорости реакции (k). Эту характеристику можно найти с помощью уравнения Аррениуса, если известны предэкспоненциальный множитель (k0), энергия активации (Е) и температура (Т):
k = k 0 e - E / RT (1.9)
Значение константы скорости можно также определить графическим способом. Так, уравнения (1.1), (1.4) и (1.6) представляют собой прямолинейную зависимость соответствующих функций концентраций от времени: при n = 1 линейно от времени зависит lnC, при n = 2 – 1/С, при n = 3 – 1/С2 (рис. 1, 2, 3). На этой закономерности основан графический метод определения порядка реакции, позволяющий также определить и значение k.
Пусть n = 1 или 2. Тангенс угла наклона (α) соответствующей прямой к оси Х (ось времени) равен константе скорости (рис 1.1, 1.2) - tgα = k. При n = 3 удвоенная константа скорости равна тангенсу угла (α) наклона прямой (рис. 1.3) - 2 tgα = k. Не представляет затруднений нахождение времени полупревращения из этих графиков.
|
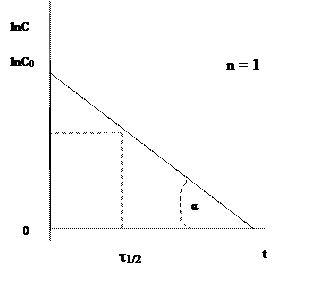
Рис. 1.1
|

Рис. 1.2
|
| |
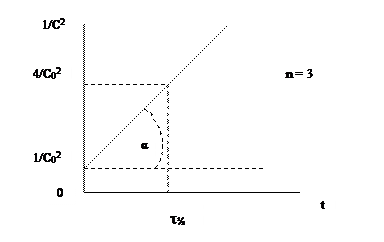
Рис. 1.3
В присутствии катализатора скорость химической реакции возрастает, что связано с изменением механизма реакции, приводящего к снижению энергии активации процесса. При катализе катализатор и реагирующее вещество образуют промежуточное соединение, которое в результате дальнейших преобразований превращается в конечный продукт, высвобождая катализатор. Энергетические затраты таких процессов ниже, чем в отсутствии катализатора. Каталитические реакции являются сложными, так как протекают в несколько стадий. Скорость процесса определяется скоростью самой медленной стадии катализа.
Величиной, определяющей ускорение реакции в при катализе, является соотношение констант скоростей реакций в присутствии (kк) и отсутствии (k) катализатора: kк/ k. Это значение можно рассчитать, если известна величина снижения энергии активации ∆Е. В расчетах будем полагать, что рассматриваемая реакция является односторонней, то есть необратимо протекает слева направо в соответствии с уравнением реакции.
Пусть ЕК = (Е - DЕ) – энергия активации прямой реакции в присутствии катализатора, Е – то же, но без катализатора. Тогда константа скорости прямой реакции в присутствии катализатора - kK = k0 exp[-(E - DE)/RT], без катализатора - k1 = k0 exp(-E/RT).
Ускорение при катализе равно:
kK/ k = exp ( D E)/RT). (1.10)
Содержание домашнего задания №1
- Расчёт константы скорости (k 1) заданной реакции при температуре (Т1).
- Построение графика 1 зависимости концентрации первого исходного вещества от времени: С i = f ( ti ).
- Определение времени полупревращения аналитическим (t 1/2ан) и графическими (t 1/2гр) методами.
- Расчёт процента превращения (pI) и массы первого исходного вещества (m 1) за определённое время (t 1). Определение масс остальных участников реакции к этому моменту времени.
- Определение температуры (Т2), при которой за данное время ((t 1) прореагирует заданная часть первого исходного вещества (p 2).
- Построение кинетического графика 2 в "спрямляющих" координатах. Определение по этому графику константы скорости реакции (k 1гр) и времени полупревращения (t 1/2гр ).
- Расчёт ускорения реакции при использовании катализатора (k 1 K / k 1).
Оформление расчета
Полагаем, что заданная реакция является простой (1-6 пункт задания) и односторонней (1-7 пункт задания).
1. Для нахождения константы скорости реакции (k 1) нужно использовать заданное значение температуры (Т1), предэкспоненциального множителя (k 0), и энергии активации (Е). Расчет выполняется по уравнению Аррениуса (1.9). Следует указать размерность величины k1 с учетом фактического (заданного) порядка реакции.
2. Для построения графика 1 зависимости текущей концентрации (С) первого исходного вещества от времени (t) необходимо составить таблицу 1.1.
В таблицу вносят рассчитанные промежуточные значений Сi, используя заданное значение начальной концентрации С0.
Далее, выбрав соответствующее заданному порядку кинетическое уравнение (1.1, 1.4, 1.6), для каждого из значений Сi нужно рассчитать значения текущего момента времени (ti) и занести их в таблицу 1.1. По данным этой таблицы (значения колонок 1 и 2) строят график 1: С = f ( t ).
При построении графика 1 (и далее графика 2) нужно помнить, что все расчетные точки соответствуют теоретическим моделям, поэтому при соединении точек должны быть образованы идеально построенные графические зависимости без выступов и изломов.
Таблица 1.1
| Сi (моль/л) | ti (с) | Линейная функция концентрации - |
| С0 = | t0=0 | |
| С1 =0,9 С0 = | t1 = | |
| С2 =0,8 С0 = | t2 = | |
| С3 =0,7 С0 = | t3= | |
| С4 =0,6 С0 = | t4= | |
| С5 =0,5 С0 = | t5= | |
| С6 =0,4 С0 = | t6= | |
| С7 =0,3 С0 = | t7= | |
| С8 =0,2 С0 = | t8= | |
| С9 =0,1 С0 = | t9= |
3. Для того, чтобы определить аналитическое значение времени полупревращения (t 1/2ан.), нужно использовать в расчете одну из формул (1.3), (1.5). (1.7), выбрав ее, в зависимости от заданного порядка реакции.
Графическое значение t 1/2гр можно найти по графику 1, как время, за которое концентрации уменьшается в два раза.
Следует рассчитать относительную ошибку (e) при нахождении времени полупревращения:
e (%) = ± [(t1/2ан – t1/2гр)/ t1/2ан] · 100 (%). (1.11)
4. Процент превращения первого исходного вещества (p1) можно определить по формуле:
p 1 = (C1пр/ С0)· 100 (%). (1.12)
Используя заданное значение времени t 1, можно найти с помощью соответствующего кинетического уравнения (1.3, 1.5. 1.6) значение текущей концентрации C1т; далее по формуле (1.8) нужно определить прореагировавшую концентрацию C1пр, затем по формуле (1.12) вычислить искомую величину p1.
Массу первого вещества (mI, г), прореагировавшего за время t 1, можно определить, используя формулу, позволяющую рассчитать соответствующую молярную концентрацию
С1пр = mI /MI·V (1.13),
где MI –молярная масса этого вещества, г/моль, V – заданный объем, л. Полученную величину mI следует перевести в килограммы.
Нужно также определить массы остальных участников реакции, прореагировавших к моменту времени t 1. Расчет делают по заданному уравнению химической реакции относительно найденной величины mI с учетом стехиометрических коэффициентов и молярных масс веществ. Например, для реакции aA +bB =lL + nN,
где mA= mI, МА=МI – молярная масса первого вещества
mB = (mI· b·MB)/(a·MI). (1.14)
Аналогичным образом можно найти массы остальных продуктов реакции - mL, mN.
Следует проверить выполняемость закона сохранения масс:
mA + mB ≡ mL,+ mN. (1.15)
5. Заданное значение степени превращения первого вещества p 2 отличается от найденной ранее величины p1, так как полагается, за одно и то же время t 1 процентное количество прореагировавшего вещества различно, из чего следует, что реакции протекают при разных температурах Т1 и Т2 и, следовательно, имеют разные константы скорости - k 1 при температуре Т1 и k 2 при температуре Т2.
Для определения температуры Т2 сначала надо найти новое значение прореагировавшей концентрации первого исходного вещества С2пр, используя формулу (1.12). Затем следует найти новое значение текущей концентрации С2т с помощью формулы (1.8).
С найденным значением С2т можно рассчитать константу скорости реакции (k2) при температуре Т2 по кинетическому уравнению реакции заданного порядка (одна из формул 1.1, 1.4, 1.6).
Определив k2, по уравнению Аррениуса (1.9), нужно рассчитать температуру Т2 (К). Для этого надо прологарифмировать уравнение (1.9) и выразить Т2:
T 2 = E/[Rln(k0/k2] (1.16)
6. В зависимости от заданного порядка реакции следует выбрать функцию концентрации, линейно зависящую от времени, рассчитать соответствующие значения и внести их в третью колонку таблицы 1.1.
Значения функции, определяют, используя величины Сi первого столбца этой таблицы. По данным второй и третьей колонок данной таблицы строят график 2.
Масштаб по оси X (ось времени, t) графика 2 можно выбрать таким же, как на графике 1, а масштаб на оси Y желательно подобрать так, чтобы наклон прямой линии был близок к 450. Это обеспечивает наибольшую точность определения константы скорости реакции по угловому коэффициенту графика 2. Для этого необходимо выделить прямоугольный треугольник и найти отношение величин вертикального и горизонтального катетов (не в единицах длины, а в единицах тех величин, которые они выражают). Полученную величину k 1гр надо сравнить с рассчитанной k1 в первом задании и найти относительную ошибку:
e (%) = ± [(k1 – k1гр )/ k1] · 100 (%). (1.17)
По графику 2 необходимо также найти время полупревращения первого вещества, t1/2гр. Для этого на оси Y следует отложить величину, соответствующую функции половинной концентрации.
Для первого порядка это будет значение ln(C0/2)=lnC0-ln2, то есть от значения lnC0 надо отложить вниз отрезок, равный ln2, через полученную точку провести горизонтальную прямую, до пересечения с графиком, и из точки пересечения опустить перпендикуляр на ось Х.
Для реакции второго порядка надо представить на оси Y величину 2/C0, что можно сделать, прибавив к отрезку, выражающему 1/C0, отрезок такой же длины, из полученной точки провести горизонтальную прямую и т.д.
Для реакции третьего порядка надо представить на оси Y величину (2/C0)2=4/C02, что можно сделать, прибавив к отрезку, выражающему 1/C02, отрезок утроенной длины.
7. Увеличение скорости реакции в присутствии катализатора следует определить по формуле (1.10).
Абсолютное значение снижения энергии активации (DE, Дж/моль) можно найти, зная величину снижения энергии активации в процентах (h,%):
DE = (h/100)·Е (1.18).
Таблица исходных данных
| Реак- ция | Порядок реакции, n | Т1, К | k0, (л/моль)n – 1с-1 | E, Дж/моль | C, моль/л | t1, c | V, л | p2, % | h |
Таблица конечных данных
| Константа скорости, k 1, (л/моль)n – 1 (с)-1 | Время полупревра-щения t1/2, с | Расход первого вещества к моменту t1 |
Т2, К | Ускоре-ние при катализe
k 1 K / k 1 | ||
| t 1/2гр | t 1/2ан | p1, % | mI , кг | |||
Вопросы для самоконтроля
1. Что такое порядок реакции, какие значения он может принимать?
2. Можно ли по уравнению химической реакции, по виду графика 1 определить порядок реакции? Ответ пояснить.
3. Что такое молекулярность реакции? Когда порядок и молекулярность реакции совпадают?
4. Дать определение времени полупревращения и пояснить нахождение этой величины с помощью графиков 1 и 2 для простых реакций 1,2 и 3 порядков.
5. Написать кинетическое уравнение реакции n-порядка. Что понимают под символом «С» в этом уравнении? При каких значениях n это выражение справедливо?
6. Какие данные ДЗ-1позволяют определить, что температура Т2 изменилась по сравнению с Т1 в правильном направлении?
7. В чем суть графического метода определения порядка реакции? О чем свидетельствует вид графика 2?
8. Причины увеличения скорости реакции в присутствии катализатора.
9. Если бы заданная реакция была двусторонней, то, как изменилась бы энергия активации обратной реакции в присутствии катализатора? Ответ пояснить.
Домашнее задание № 2. «Химическая термодинамика»
Элементы теории
Химическая термодинамика даёт возможность рассчитать важнейшие количественные характеристики химических реакций — теплоту (q,), выделяемую или поглощаемую в результате проведения реакции, максимальную работу (A), которую можно получить при проведении реакции, а также константу равновесия (Kp) реакции и зависимость этой константы от температуры.
Уравнения химической термодинамики выводятся из двух постулатов - первого и второго начал термодинамики. Эти уравнения включают набор величин, называемых функциями состояния, которые характеризуют равновесное состояние термодинамической системы. Функции состояния включают как простые для понимания и измеримые величины (параметры) - объём (V), температуру (Т), давление (Р), теплоёмкость при постоянном давлении (Ср) и при постоянном объёме (Сv), так и более сложные — внутреннюю энергию (U), энтальпию (Н), энтропию (S), энергию Гиббса (G) и энергию Гельмгольца (F).
Энтальпия равна сумме внутренней энергии и произведению давления на объём системы, то есть
Н = U + PV. (2.1)
Для веществ в конденсированном состоянии энтальпия практически не отличается от внутренней энергии. Энтальпию используют при описании изобарных процессов.
Энергия Гиббса выражается комбинацией функций состояния:
G = U + PV – TS = H – TS, (2.2)
Энергия Гельмгольца равна:
F = U – TS (2.3)
Известны значения измеримых характеристик (например, давление, объем) и энтропии, а абсолютные значения U, H, G и F неизвестны. Это обстоятельство не препятствует их использованию для расчётов практически важных величин, так как необходимо знать лишь их изменения в различных процессах, а эти изменения можно определить экспериментально.
Уравнения химической термодинамики включают также теплоту и работу. Эти величины не являются функциями состояния, это формы передачи энергии между термодинамической системой и окружающей средой: теплота - посредством неупорядоченного движения частиц, работа - посредством упорядоченного движения частиц.
Конечное изменение (то, которое можно измерить) функции состояния обозначают греческой буквой дельта - D, бесконечно малое изменение обозначают знаком дифференциала - d. Конечное изменение характеристик, не являющихся функциями состояния, принято обозначать соответствующими буквами, например А - конечное изменение работы; бесконечно малое изменение таких характеристик будем обозначать буквой d.
Работа условно делится на две части, одна часть — работа расширения против внешнего давления – А расшир. (она при постоянном давлении равна PDV), остальная часть называется полезной работой – А пол. (это название не имеет буквального смысла):
А = А пол. + А расшир. (2.4)
Тепловым эффектом реакции называется количество теплоты, выделяемое или поглощаемое в результате проведения химической реакции, при постоянной температуре и при условии, что А пол. = 0, то есть совершается только работа расширения. При этих условиях из первого начала термодинамики выводится соотношение для теплового эффекта реакции:
при V=const Qv = ∆U, при P=const Qp = ∆ Н, (2.5)
где Qv, Qp- тепловой эффект реакции при постоянном объеме и постоянном давлении соответственно.
Эти формулы выражают закон Гесса, согласно которому тепловой эффект реакции не зависит от пути проведения реакции, а зависит лишь от начального и конечного состояния термодинамической системы. Таким образом, измерив тепловой эффект реакции, можно определить величину изменения внутренней энергии и энтальпии в результате реакции. Реакции, протекающие с выделением тепла, называются экзотермическими (∆ U <0, ∆Н<0), реакции, протекающие с поглощением тепла, называются эндотермическими (∆ U >0, ∆Н>0).
Закон Гесса имеет два следствия, фактически это две формулы для расчёта теплового эффекта реакции.
В первом случае тепловой эффект данной реакции рассматривается как алгебраическая сумма тепловых эффектов двух мысленно проводимых реакций: I) «разборка» молекул исходных веществ на элементарные «кирпичики» - наиболее устойчивые группировки атомов элементов - «простые вещества»; 2) «сборка» молекул продуктов из «кирпичиков» - простых веществ.
Тепловой эффект сборки молекул из простых веществ при стандартных условиях (1 атм, 298 К) называется стандартной теплотой образования вещества (∆fH0) и измеряется в Дж/моль. На стандартность указывает верхний индекс 0 при написании соответствующей термодинамической величины.
Таким образом, тепловой эффект реакции при стандартных условиях (DH0) равен разности сумм стандартных теплот образования конечных (индекс j) и исходных (индекс j) продуктов с учетом стехиометрических коэффициентов реакции (ni, ni):
DH0 = S (nj DfH0j)кон.. - S(n i DfH0i)исх. (2.6)
Во втором следствии тепловой эффект данной реакции рассматривается как алгебраическая сумма тепловых эффектов двух мысленно проводимых реакций:
1) окисление молекул исходных веществ кислородом с образованием высших оксидов элементов, из которых состоят исходные вещества;
2) реакция, обратная окислению молекул продуктов. Тепловой эффект реакции окисления для 1 моля вещества при стандартных условиях называется стандартной теплотой сгорания (∆сгH0, Дж/моль).
В этом случае тепловой эффект реакции при стандартных условиях (DH0) равен разности сумм стандартных теплот сгорания исходных и конечных продуктов с учетом стехиометрических коэффициентов реакции:
DH0 = S (ni DсгH0i)исх.. - S(nj DсгH0j)кон.. (2.7)
Справочные данные по теплотам образования или сгорания веществ относятся к стандартным условиям, что позволяет рассчитать тепловой эффект реакции только при стандартной температуре 298°К.
Для расчётов теплового эффекта при других температурах необходимо привлечь теплоёмкости веществ. Теплоёмкость при постоянном объёме равна производной внутренней энергии по температуре - Cv = dU/dT, а теплоёмкость при постоянном давлении равна производной энтальпии по температуре - Ср = dH/dT. Размерность теплоемкости – Дж/(моль∙К).
Наиболее часто используют величину Ср. Для Ср различных веществ накоплены экспериментальные данные и предложены эмпирические уравнения, с помощью которых можно рассчитать теплоёмкость при любой заданной температуре - для неорганических веществ :
Ср = а + bТ + с/Т-2, (2.8)
для органических веществ:
Ср = а + bТ + сТ2 -. (2.9)
Коэффициенты a, b, c/, c находят опытным путем и включают в справочники.
Тепловой эффект реакции при различных температурах можно рассчитать с помощью закона Кирхгофа, в соответствии с которым в изобарном процессе температурный коэффициент теплового эффекта химической реакции - d(DH)/dT равен изменению теплоемкости системы в результате реакции – DСp:
d(DH)/dT = Dcp. (2.10)
Для нахождения расчетной формулы нужно разделить переменные и проинтегрировать в пределах интервала температур от стандартной 298 до заданной - Т (К). Получим выражения для двух случаев: 1) в реакции участвуют неорганические вещества; 2) в реакции участвуют неорганические и органические вещества. Полагаем, что реакции протекают при стандартном давлении. В результате приходим к следующим выражениям:
DH0T = DH0+Da∙(T-298) + (Db/2)∙(T2 -2982) - Dс/∙(1/T-1/298), (2.11)
DH0Т = DH0+Da∙ (T-298)+(Db/2)∙(T2-2982) - Dc/∙(1/T-1/298)+(Dc3/3)∙(T3-2983). (2..12)
В этих формулах DH0T – тепловой эффект реакции при стандартном давлении и температуре Т, DH0 – тепловой эффект реакции при стандартных условиях; Da, Db, Dc , Dc/∙ - изменение соответствующих коэффициентов a, b, c , c/∙ в формулах для температурной зависимости теплоемкости (2.8, 2.9) по всем участникам реакции:
Da = S(nj aj)кон.. - S(ni ai)исх., (2.13)
где ni, ni - стехиометрические коэффициенты; значения Db, Dc, Dc/ рассчитывают аналогично.
Второе начало термодинамики позволяет предсказать, в каком направлении пойдёт процесс, если он протекает самопроизвольно. Энтропия вещества - мера неупорядоченности на микроуровне, это столь же важная термодинамическая характеристика вещества, как и внутренняя энергия. Измерить энтропию невозможно, но её можно рассчитать следующим образом. По определению, приращение энтропии (dS) равно приведенной теплоте процесса поглощения системой тепла, то есть отношению поглощенного тепла δq к абсолютной температуре: dS = δq/T. Эта формула справедлива только в том случае, если теплота поступает достаточно медленно, в обратимом равновесном процессе.
Третье начало термодинамики утверждает, что при температуре абсолютного нуля энтропия идеального кристалла равна 0. В изобарном процессе dS = CpdT/T. Интегрирование этого выражения в пределах изменения температуры от 0 К до 298 К позволяет рассчитать абсолютное значение энтропии S0 Эта величина называется стандартной абсолютной энтропией вещества, измеряется в Дж/(моль К) и её значение для каждого вещества можно найти в справочниках по термодинамике.
Энтропия смеси веществ равна сумме энтропий составляющих её компонентов, а изменение энтропии в результате реакции (DS) рассчитывается как разность сумм энтропий конечных продуктов и исходных веществ с учетом стехиометрических коэффициентов реакции:
DS = å(nj Sj)кон. - å(ni Si)исх . (2.14)
Для стандартных условий в расчёты входят стандартные абсолютные энтропии веществ - участников реакции, а для произвольно заданной температуры необходимо рассчитать энтропии веществ при этой температуре с помощью теплоёмкостей. Так, для реакций, протекающих при температуре Т, отличной от 298 К, между неорганическими веществами (1) и для реакций между неорганическими и органическими веществами (2) соответствующие формулы приведены ниже:
DSТ0 = DS0 +Da ln(T/298) +Db∙ (T-298) – (Dc//2)∙ (1/T2–1/2982), (2.15)
DSТ0= DS0 + Da∙ ln(T/298) +Db∙ (T-298) – (Dc/ /2)∙(1/T2 – 1/2982) + (Dc /2)∙ (T2 – 2982). (2..16)
В этих формулах DS0 – изменение энтропии реакции при стандартных условиях, DSТ0 – изменение энтропии реакции при температуре Т и стандартном давлении; Da, Db, Dc, Dc/ - те же величины, что в формулах (2.11, 2.12).
Энергия Гиббса системы ранее называлась изобарно-изотермическим потенциалом. Это одна из наиболее применяемых термодинамических функций системы, с её помощью можно:
- предсказать направление самопроизвольно протекающего процесса (P,T=const): процесс пойдёт в сторону уменьшения G;
- рассчитать полезную максимальную работу, которую можно получить в результате проведения реакции - А пол ≤ -∆ G .
- найти константу равновесия реакции с помощью уравнения химического сродства - Кр = exp (- ∆ G °/ RT ) (2.17).
Химическое сродство характеризует способность веществ взаимодействовать друг с другом. Мерой химического сродства является величина ∆G0 – изменение энергии Гиббса в стандартных условиях. Чем эта величина меньше, тем эффективнее вещества взаимодействуют друг с другом.
Величина Kp характеризует химическое равновесие. Ее можно рассчитать не только с помощью уравнения (2.17), но и из закона действующих масс. Так, в гомогенной системе (реагирующие вещества находятся в одной фазе) Kp равна отношению произведения равновесных парциальных давлений конечных продуктов к произведению равновесных парциальных давлений исходных продуктов, взятых в степенях, равных соответствующим стехиометрическим коэффициентам. Kp зависит от температуры и природы реагирующих веществ.
Уравнение Вант-Гоффа:
dlnKp/dT = DH/RT2 (2.18)
где DH – тепловой эффект реакции, позволяет предсказать, в какую сторону будет изменяться константа равновесия при изменении температуры. Если реакция эндотермичная (DH>0), то с ростом температуры константа будет расти, если реакция экзотермичная – наоборот.
Дата добавления: 2020-11-27; просмотров: 58; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!