Наследственные дефекты метаболизма (общая характеристика, классификация)
Это группа заболеваний, связанных с расстройством метаболизма, иначе их название звучит «врожденные ошибки обмена». Их развитие - это следствие дефекта единичных генов, обеспечивающих трансформацию определенных веществ. Большинство наследственных болезней обмена передается по рецессивному типу и крайне редко по доминантному.
Классификация:
1. Нарушение обмена аминокислот;
2. Нарушение обмена органических кислот и углеводов;
3. Болезни накопления (мукополисахаридозы, липидозы, каридозы);
4. Лейкодистрофии (нарушение структуры белого вещества);
5. Обменно-гормональные нарушения синтеза гормонов;
6. Другие нарушения обмена веществ.
Существует перечень общих признаков, указывающих на патологию обмена веществ:
· судорожный синдром;
· отказ от пищи с прогрессирующей потерей веса;
· рвота;
· специфический запах тела и мочи;
· длительная желтуха;
· поражение мышц;
· увеличение размеров печени;
· задержка физического развития.
Фенилкетонурии (общая характеристика)
Это заболевание развивается вследствие нарушения обмена веществ, точнее, фенилаланина — ароматической альфа-аминокислоты, которая накапливается в организме ребенка и приводит к токсическому поражению центральной нервной системы и значительному отставанию малыша в развитии.
Подобные болезни обмена веществ у детей выявляются достаточно редко, так как они сложны в диагностике и только ближе ко второму полугодию жизни возникают явные признаки их развития.
Фенилкетонурия 1-го типа
Этиология
Заболевание наследуется аутосомно-рецессивно и вызвано мутацией, которая вызывает снижение активности фермента фенилаланин-4-монооксигеназы, обеспечивающей превращение фенилаланина в тирозин. Фермент имеется только в печени, почках, поджелудочной железе.
Эпидемиология
Частота — 1:10000 новорожденных.
Клиническая картина
Первый признак — рвота, ребенок становится вялым, сонливым, не проявляется интерес к окружающему, раздражителен, беспокоен, плаксив, срыгивает, нарушение мышечного тонуса, судороги, повышенная потливость, «мышиный запах», отстают в физическом и нервно-психическом развитии
Продолжительность жизни в норме.
Мукополисахаридозы
Группа редких генетически заболеваний, причиной которых является нехватка определенных ферментов, которые помогают расщеплять определенные виды жиров и углеводов на простые молекулы.
Наследуется по аутосомно-рецессивному типу.
Выявляется непропорционально малый рост, задержка которого начинается к концу первого года жизни. Грубые черты лица: нависающий лоб, большой язык, гипертелоризм, деформация ушей и зубов. Грудная клетка деформирована, выражен кифоз (искривление назад) грудного и поясничного отделов позвоночника. Характерны ограничение подвижности суставов, увеличение селезенки и печени одновременно, пупочные и паховые грыжи. Раннее окостенение затылочно-теменного шва, «рыбьи позвонки» (позвонки как двояковогнутые линзы). Снижение мышечного тонуса, общая двигательная заторможенность. Снижение интеллекта и ослабление слуха разной степени характерны для различных типов мукополисахаридозов.
Мукополисахаридоз 1-го типа
Этиология
 Аутосомно-рецессивное заболевание, обусловленное мутациями в структурном гене фермента альфа-L-идуронидазы, который расположен на коротком плече хромосомы 4.
Аутосомно-рецессивное заболевание, обусловленное мутациями в структурном гене фермента альфа-L-идуронидазы, который расположен на коротком плече хромосомы 4.
Эпидемиология
Частота — 1:100000 новорожденных.
Продолжительность жизни
Быстро прогрессирующее течение, приводящее к смертельному исходу на первом десятилетии жизни в результате сердечно-легочных и неврологических нарушений.
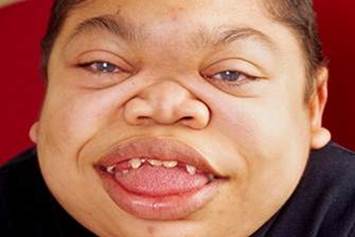 43. Мукополисахаридоз 2-го типа
43. Мукополисахаридоз 2-го типа
Этиология
В основе лежат мутации Х-сцепленного гена идуронатсульфатазы, кодирующего одноимённый лизосомный фермент.
Эпидемиология
Частота — 1:140000-156000 новорожденных.
Клиническая картина
Соматические нарушения: макроцефалия, грубые черты лица, низкий рост, короткая шея, аномалии зубов, толстая кожа, атипичный пигментный ретинит (воспаление сетчатки) или разрушение сетчатки, грыжи, тугоподвижность суставов.
Внутренние органы: заболевания дыхательных путей, сердца
НПР: прогрессирующая проводящая или нейросенсорная потеря слуха, поражение ЦНС.
Продолжительность жизни
Дети с тяжелой формой погибают в конце второй декады жизни. Больные с легкой формой могут жить 50-60 лет.
Мукополисахаридоз 3-го типа
Этиология
Обусловлен дефицитом разных ферментов, но во всех случаях в лизосомах накапливается один тип гликозаминогликанов – гепарансульфат.Аутосомно-рецессивный тип наследования.
Эпидемиология
Частота — 1:70000 новорожденных.
Клиническая картина
Соматические нарушения: жесткие волосы, гирсутизм(рост волос по мужскому типу), отставание в росте, легкие скелетные изменения.
Внутренние органы: увеличение печени и селезенки, пороки развития сердца
НПР: УО, регресс психомоторного и речевого развития, гиперактивны, аутичны или агрессивны, нарушен сна, небрежны и невнимательны, судороги.
Продолжительность жизни
Умирают в возрасте до 30 лет.
Дата добавления: 2019-11-25; просмотров: 336; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!