БИОХИМИЧЕСКИЕ ОСНОВЫ АЛКАПТОНУРИИ
А. С. Долгова, Д. С. Мастюкова, А. А. Лободина, 2 курс
Научный руководитель - к.м.н., доц. С. Н. Афонина
Кафедра биологической химии
Оренбургский государственный медицинский университет
BASIC BIOCHEMICAL ALKAPTONURIA
A.S. Dolgova A.A.Lobodina D.S. Mastukova, 2 course
Candidate of Sciences in Medicine, Assistant Professor S.N. Afonina
Department of Biochemistry
The Orenburg State Medical University
Аннотация. На сегодняшний день, такая патология как, алкаптонурия диагностируется ещё в ранннем возрасте. Однако в некоторых случаях она может быть выявлена только по мере развития полноценной клинической картины. Несмотря на достижения в медицине и биохимии в борьбе и профилактике алкаптонурии, проблема ее лечения остаётся актуальной.
Ключевые слова: алк a птонурия, биохимия, лечение, профилактика.
Annotation. Today, alcaptonuria is diagnosed at an early age. Sometimes it can be detected, if develops the full clinical picture. Despite advances in medicine and biochemistry in the treatment and prevention of alkaptonuria, the problem of its treatment remains relevant.
Key Words: alkaptonuria, biochemistry, treatment, prevention.
Алкаптонурия — генетически обусловленная энзимопатия, в основе которой лежит нарушение метаболизма тирозина, приводящее к избыточному образованию промежуточного метаболита - гомогентизиновой кислоты (HGD), определяющая течение симптомокомплекса алкаптонурии.. В последние годы частота выявления данной энзимопатии растёт, что обусловливает повышение необходимости её исследования.
Цель статьи. Рассмотреть биохимические основы алкаптонурии. Для её достижения нами поставлены задачи: определить нарушение в обмене тирозина и выявить характерные для него симптомы и осложнения.
|
|
|
Материалы и методы. Анализ материалов, посвящённых патологии обмена тирозина.
При алкаптонурии мутации затрагивают ген оксидазы HGD, поэтому нарушается воспроизводство фермента гомогентизиназы, участвующей в расщеплении тирозина и фенилаланина.
Гомогентизиновая кислота при данной патологии превращается в хиноновые полифенолы (пигмент алкаптон), которые накапливаются в соединительных тканях и экскретируются с мочой.
Клиническую картину алкаптонурии составляют гомогентизиновая ацидурия, охроноз и артропатия. Существует и ранний признак - в детском возрасте на мокрых пеленках из-за большого количества гомогентизиновой кислоты в моче от нее остаются темные разводы, однако полный симптомокомплекс развивается к 20-35 годам. В дальнейшем со стороны мочеполовых органов нередко развиваются пиелонефрит, мочекаменная болезнь, калькулезный простатит.
Кожный синдром при алкаптонурии характеризуется появлением серо-коричневой пигментации, уплотняются и приобретают серо-голубое окрашивание ушные раковины. Диффузное отложение пигмента отмечается в хрящах гортани, что сопровождается охриплостью голоса, дисфагией и болью при глотании. Кроме того, патология затрагивает крупные суставы нижних конечностей и позвоночник.
|
|
|
Таким образом, несмотря на лёгкость постановки диагноза, на сегодняшний день специфического лечения указанной патологии не существует. Это вызывает необходимость разработки принципиально новых подходов в лечении алкаптонурии.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ ТИРОЗИНЕМИИ I ТИПА
Д.А. Козедуб, 2 курс
Научный руководитель – к. б. н., доц. И.В. Карнаухова
Кафедра биологической химии
Оренбургский государственный медицинский университет
ETIOLOGY AND PATHOGENESIS OF TYROSINEMIA TYPE I
D.A. Kozedub, 2 course
Supervisor – Candidate of Biological Sciences, docent I. V. Karnauhova
Department of Biological Chemistry
The Orenburg State Medical University
В данной статье ставится задача рассмотреть этиологию и патогенез тирозинемии Iтипа. При анализе источников были выделены основные формы заболевания, их клиническая картина и осложнения. Особое внимание обращается на методы лечения патологии, которые обеспечивают длительную выживаемость больных.
Ключевые слова: тирозинемия, фумарилацетогидролаза, деградация тирозина, митихондриальные токсины, печеночная дисфункция, формы заболевания, тубулопатия, печеночная недостаточность, порфирии, нитизинон, диетотерапия, трансплантация печени.
|
|
|
This article aims to examine the etiology and pathogenesis of tyrosinemia type I. When analyzing the sources, were identified the main forms of the disease, their clinical picture and complications. Particular attention is drawn to the methods of treatment of pathology, which provide long-term survival of patients.
Key words: tyrosinemia, fumarylacetohydrolase, tyrosine degradation, mitochondrial toxins, hepatic dysfunction, forms of the disease, tubulopathy, liver failure, porphyrias, nitizinon, diet therapy, liver transplantation.
Тирозинемия–заболевание, связанное с дефицитомактивности фумарилацетоацетатгидроксилазы, приводящее к нарушению метаболизма тирозина.На сегодняшний день выделяют 3 типа данного заболевания.
Наследственная тирозинемия I типа (гепаторенальная тирозинемия) – редкое генетическое заболевание с аутосомно-рецессивным типом наследования (25% сибсов больного могут страдать данной патологией, 50-% могут быть здоровыми носителями, 25% - здоровыми и не являться носителями болезни), обусловленное мутациями в гене фермента фумарилацетоацетазы (фумарилацетогидролазы (FAH)). Ген FAHрасположен на длинном плече 15-й хромосомы.
В большом количестве FAHсодержится в гепатоцитах,а также клетках почечных канальцев.
Заболевание впервые описал M. Barberв 1956 году. Распространено во всех странах мира со средней частотой – 1:100 000 новорожденных.Частота носительства мутаций НТ 1 в популяциях – 1:150 человек. В Квебеке (Канада) 1 из 20 жителей является носителем мутантного гена.
|
|
|
В норме фумарилацетогидролаза осуществляет конечный этап деградации тирозина на фумарат и ацетоацетат (нетоксичны). В результате дефекта мутации белка, кодирующего FAH, распад тирозина происходит по альтернативному патологическому пути. В результате образуются высокотоксичные и канцерогенные сукцинилацетон, малеилацетоацетат, фумарилацетоацетат.
Патогенез заключается в интоксикации выше перечисленными продуктами аномального распада тирозина, которые являются митохондриальными токсинами. Они тормозят фосфорилирование и блокируют цикл Кребса. Первым пораженным органом является печень.
В течение первых месяцев жизни проявляется печеночная дисфункция, переходящая в почечную недостаточность, цирроз, ренальные тубулопатии, гипофосатемический рахит,синдром Фанкони (почечный тубулярный ацидоз, гипофосфатемия и аминоацидурия)и печеночную карциному (40% детей). Может развиться нефрокальциноз и почечная недостаточность.
Выделяют острую, подострую и хроническую формы.
Острая форма (HT1А) развивается у детей в первые месяцы жизни и встречается у 75% больных. Протекает по типу быстропрогрессирующей печеночной недостаточности и заканчивается смертью ребенка. Часто НТ 1А не распознается или подтверждается уже после смерти ребенка. Признаки поражения почек выражаются в развитии тубулопатий, которые, вследствие нарушения обмена кальция и фосфора, могут приводить к рахитоподобным проявлениям. Увеличение уровня тирозина и метионина в сыворотке вызывают появление «капустного» запаха от тела и мочи пациента. Данный тип заболевания сопровождается задержкой развития, рвотой, анемией, гематурией, диарей или динамической непроходимостью, гепатомегалией. Смерть наступает в результате печеночной недостаточности или кровотечения, также встречается отек легких и мозга. Смерть 90% больных регистрируется в возрасте до 2 лет.
Подострая форма характерна для детей во втором полугодии жизни. Характеризуется задержкой в развитии, гепато- или гепатоспленомегалией, печеночной недостаточностью, циррозом, задержкой в развитии. Могут появляться параличи конечностей и диафрагмы, желтуха.
Хроническая форма (НТ 1Б) характерна для детей старшего возраста и подростков. НТ 1Б характеризуется более поздним и медленным развитием. Проявляются поражения печени, рахитоподобные изменения скелета (остеопороз и остеомаляция), кардиомиопатия, неврологические кризы (1-7 дней), гипогликемия, гиперсекреция инсулина. Заболевание может начаться с непеченочных проявлений по типу порфирии (периферической нейропатии, острыми абдоминальными кризами). Без лечения летальный исход наступает примерно в 10 лет.
Диагноз базируется на высоком уровне тирозина в плазме крови (часто наблюдается гиперфенилаланинемия) и метаболитах гидроксифенилаланина в плазме и моче (гидроксифенилуксусная и гидроксифенилмолочная кислоты).
На сегодняшний день этиотропная и ферментозаместительная терапия данного заболевания не разработаны.
В последнее время все более широкое применение получает нитизинон (орфадин), используемый в качестве патогенетической терапии. Это препарат, ингибирующий 4-гидрооксифенилпируватдиоксигеназу – фермент, участвующий на втором этапе метаболизма тирозина и обеспечивающий образование токсичных метаболитов тирозина. Конечно, чем раньше пациент начнет использовать данный препарат, тем больше шансов обойтись без трансплантации печени и развития гепатокарциномы.
До разработки патогенетической терапии диетотерапия играла ведущую роль в лечении заболевания. Суть диетотерапии заключается в ограничении поступления белка с пищей, тирозина, метионина, фенилаланина.
Трансплантация печени рекомендуется при прогредиентном течении патологического процесса и обеспечивает длительную выживаемость 85% больных.
Своевременное лечение дает положительные результаты, поэтому важно с помощью скринирующих программ выявить дефект до появления клинических симптомов.
A NEONATAL DEATH DUE TO MEDIUM-CHAIN ACYL-COA DEHYDROGENASE (MCAD) DEFICIENCY
V. Minakova,
Associate Professor, PhD
Biochemistry Department,
Saint James School of Medicine,
Saint Vincent and the Grenadines
Annotation. MCAD deficiency, the most common inherited defect of fatty acid oxidation, results in arrest of b-oxidation at the 12-carbon stage with resultant hypoglycemia, hyperammonemia, increased triglyceride synthesis, impaired ketogenesis, and the build-up of organic acids in the urine.
Key words: fatty acids oxidation, medium-chain acyl-CoA dehydrogenase (MCAD), MCAD deficiency
A male infant born at 39 weeks of gestation via cesarean section and his mother were sent home 35 hours after delivery. At home, at 61 hours of age, the infant was found unresponsive and cyanotic. During this period, the infant had received only two feedings and he slept most of the time. Paramedics were called, and the infant was transported by ambulance to the local hospital. Despite efforts at resuscitation, the infant was pronounced dead. A forensic examination did not reveal any foul play or accidental asphyxia. The only abnormality found on autopsy was extensive microvesicular metamorphosis of the liver. The infant’s heel-stick blood had been collected on a filter paper before the discharge from the hospital for the detection of inborn errors of metabolism. The specimen had been sent to a regional laboratory. The results of this metabolic profile were made available subsequent to autopsy examination and indicated MCAD deficiency. The profile showed abnormal elevation of C6, C8, and C10 fatty acyl carnitine esters. The autopsy blood acyl carnitine profile confirmed the MCAD diagnosis. The parents were carriers for two different MCAD gene mutations.
MCAD deficiency is the most common inherited autosomal recessive metabolic defect of fatty acid oxidation. Its prevalence is between 1:8000 and 1:15,000. The gene of MCAD has been mapped to locus 1p31; more than 80 allelic variations have been reported [6]. The most common mutation is 985A>G, which refers to a substitution of a guanine nucleotide for an adenine nucleotide at the 985th residue.
What are the reasons for the clinical and laboratory findings exhibited by this patient?
Many tissues (especially heart and skeletal muscle) rely heavily on fatty acid oxidation as the primary fuel source for ATP production during fasting or during times of metabolic stress, such as exercise or illness, especially illness that results in decreased oral intake. In addition, a substantial amount of these fatty acids undergo b-oxidation in the liver, which uses the resulting acetyl-CoA to produce ketone bodies that are released to provide energy for the brain (Fig. 1).
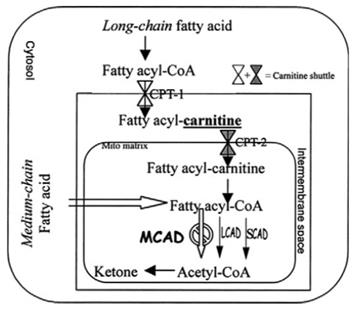
Figure 1.Summary of metabolism of fatty acids.
When MCAD is deficient, medium-chain fatty acyl-CoA molecules are unable to undergo b-oxidation in these tissues. In addition, although long-chain fatty acyl-CoA compounds can be oxidized into medium chain acyl-CoA molecules, b-oxidation is arrested at the 12-carbon fatty acyl-CoA stage. As a result, medium-chain fatty acyl CoA molecules accumulate in the cytosol and mitochondrial matrix. Some of these medium-chain compounds are converted to the organic acid derivatives as dicarboxylic acids that can be detected in the urine. The urinalysis and detection of dicarboxylic acids are considering as confirmed tests .
These derivatives may also be toxic to tissues that carry out b-oxidation.
In the liver, a substantial portion of these medium-chain fatty acyl CoA molecules is used in cytosolic re-synthesis of triglycerides, resulting in liver enlargement from fatty infiltration.
MCAD deficiency also causes ATP production and ketogenesis to be greatly decreased. Without an adequate supply of energy, the rate of the urea cycle is decreased, leading to hyperammonemia.
The rate of gluconeogenesis is similarly reduced, and the liver glycogenis rapidly exhausted, resulting in hypoglycemia.
Loss of MCAD function also results in decreased production of acetyl-CoA, which, in turn, leads to decreased ketone production despite the hypoglycemia.
This distinctive hypoketotic hypoglycemia pattern is characteristic of MCAD deficiency.
Other clinical features of MCAD deficiency reflect the involvement of organs that are dependent on b-oxidation. These findings include liver damage and liver function test (LFT) elevation; muscle enzyme elevation, hypotonia, and rhabdomyolysis; congestive heart failure; and neurologic impairment and cerebral edema.
How should this child be treated?
Avoidance of fasting and of medium-chain fatty acids in the diet is essential. Frequent small meals high in carbohydrate and protein and low in fat (<20% of calories) are recommended. Patients with MCAD deficiency should take carnitine supplements to promote efficient long-chain fatty acyl-CoA transport into the mitochondria.
Affected individuals require close monitoring of growth to make appropriate dietary changes. A skilled nutritionist should direct such changes.If the condition remains stable, blood studies beyond those required in routine pediatric care are not needed.
ГЛУТАРОВАЯ АЦИДУРИЯ I ТИПА
А.В Савельева, 6 курс
Научный руководитель – к.б.н., доц. Е.Н. Лебедева
Кафедра биологической химии
Оренбургский государственный медицинский университет
GLUTARIC ACIDURIA TYPE I
A.V. Savelieva, 6 course
Supervisor – Candidate of Biological Sciences, Assistant Professor E.N. Lebedeva
Department of biological chemistry
The Orenburg State Medical University
Одной из энзимопатий белкового обмена является глутароваяацидуриятипа 1 (недостаточностьглутарилКоАдегидрогеназы, глутароваяацидемиятип 1) аутосомнорецессивноезаболевание, обусловленноемутациямивгене,кодирующемферментглутарилКоАдегидрогеназу (GCDH).
Ключевые слова: глутароваяацидурия, энзимопатии, белковый обмен.
One of the enzymes of protein metabolism is glutaricaciduria type 1 (deficiency of glutaryl COA dehydrogenase, glutaricacidemia type 1) autosomal recessive disease caused by mutations in the gene encoding the enzyme glutarilcoa dehydrogenase (GCDH).
Keywords: glutarovyaciduria, enzymopathies, protein metabolism.
Дефицит данного ферментаприводит к накоплению в биологических жидкостях и тканях глутаровойи -3 гидроксиглутаровой, кислот, оказывающих нейротоксическое действие преимущественнона подкорковые структуры головного мозга. Диагноз глутаровойацидурии типа 1 не являетсяклиническим, он устанавливается при обнаружении определенных биохимических и/илимолекулярно генетических изменений, выявляемых у пациентов с определенной клиническойсимптоматикой или на доклинической стадии при проведении массового скринингановорожденных.
Ген GCDH картирован на хромосоме 19p13.2. К настоящему времени в гене GCDHописано более 200 различных мутаций, одной из самых распространенных является R402W,встречающаяся с частотой 12- 40% в странах Западной Европы. Некоторые мутациихарактерны для определенных этнических групп и изолятов: мутации P248L и E365K вТурции, мутации IVS 1 +5G>Tи A421V в общинах амишей.Фермент глутарилКоАдегидрогеназа (GCDH) участвует в метаболизме аминокислотлизина, гидролизина, триптофана. GCDH локализована в матриксе митохондрий ипредставляет собой гомотетрамер. Каждая субъединица этого белка состоит из 438аминокислот, N терминальный остаток (44 аминокислоты) удаляется после транспортировкифермента в митохондрию. GCDH катализирует 2 последовательных реакции превращенияглутарилКоА в кротонилКоА (реакции дегидрирования и декарбоксилирования). Врезультате блокирования данной ферментной реакции происходит накопление глутаровой иоксиглутаровой кислот в биологических жидкостях и тканях (рис.1).
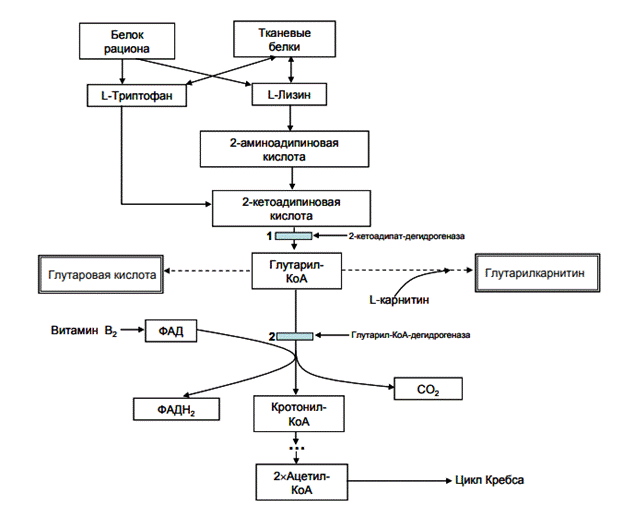
Рис.1 Схема метаболических процессов, приводящих к развитию глутаровойацидурии тип: метаболические блоки выделены голубым цветом, патологические метаболиты выделены двойной рамкой.
Мутантный фермент относится к группе флавиновыхдегидрогеназмитохондриального матрикса, участвующих в переносе электронов к убихинону в дыхательной цепи митохондрий.
Механизмы развития ГА 1 до конца не изучены. Преимущественное поражениестриарной системы связывают с избирательной токсичностью глутаровой кислоты и/или ее производных. Также глутаровая кислота и ее метаболиты ингибируют декарбоксилазуглутаминовой кислоты (фермент, участвующий в метаболизме ГАМК), что вызываетснижение концентрации этого тормозного нейромедиатора. У больных с ГА 1 выявленозначительное снижение декарбоксилазной активности в лобных отделах коры головногомозга, хвостатых ядрах и скорлупе и снижение концентрации ГАМК в цереброспинальнойжидкости (ЦСЖ). Однако данные биохимические нарушения могут быть вторичными ивозникать вследствие повреждений ГАМК эргических нейронов мозга.
Механизмы патогенеза острых «энцефалитоподобных» кризов окончательно не ясны. Считают, что глутаровая и оксиглутаровая кислоты, имеющие структурное сходство с глутаматом, взаимодействуют с N метиласпартатными рецепторами, для которых глутамат является естественным активатором, что вызывает чрезмерное накопление ионов Са2+впостсинаптических нейронах и приводит к гибели клеток. Другим возможным нейротоксическим фактором считают накопление промежуточного метаболита обмена триптофана и лизина квинолиновой кислоты. В настоящее время, до конца не распознана причина лобно височной атрофии и/или гипоплазии и субдуральных кровоизлияний при ГА1.
Считается, что во время эмбриогенеза 3 оксиглутаровая кислота может нарушать формирование стенок сосудов, в первую очередь сосудов головного мозга, приводя к повышению их проницаемости и возникновению кровоизлияний.
При аутопсии у всех пациентов с ГА1 выявляют выраженную субкортикальную и кортикальную атрофию головного мозга, атрофическую вентрикуломегалию. В большинстве случаев визуализируются некротические изменения в области скорлупы и головки хвостатого ядра. Реже поражаются зрительные тракты, мозолистое тело, внутренняя капсула, глубокие отделы белого вещества мозжечка, ствола мозга. У ряда пациентов обнаруживают «губчатую» дегенерацию белого вещества, преимущественно в перивентрикулярных областях, реже в таламусе, бледном шаре и стволе головного мозга. У большинства пациентов при аутопсии печени выявляется жировая инфильтрация клеток печени, проксимальных канальцев почек и миокардиоцитов, что может быть связано с неспецифической митохондриальной дисфункцией.
Эпидемиология. Глутароваяацидурия тип 1 (ГА1) относится к числу редких наследственных болезней обмена веществ. Частота заболевания в странах Западной Европы составляет в среднем 1:50000 живых новорожденных. Высокая частота встречаемости ГА1 отмечена среди общин амишей в Америке, Канаде и Пенсильвании 1:300. К настоящему времени в литературе имеются описания более 400 случаев этого заболевания.
Диагностика основана на данных анамнеза родословной, анамнеза заболевания,клинических симптомах, результатах лабораторных и инструментальных исследований,данных молекулярной (ДНК) диагностики. Из биохимических методов можно отметить определение в биологических жидкостях (моча и кровь) концентрации органических кислоти/или ацилкарнитинов (глутарилкарнитина). При данном заболевании повышаются
концентрацииглутаровой, 3-ОН-глутаровой кислот и глутарилкарнитина в десятки раз по
сравнению с нормой. Органические кислоты определяют методом хроматомасс-спектрометрии, ацилкарнитины - методом тандемной масс-спектрометрии.В последние годы в связи с широким внедрением тандемной-масс спектрометрии (ТМС)во многих странах проведены пилотные исследования по неонатальному скринингу наГА1.С помощью ТМС определяют концентрации глутарилкарнитина (С5DC) в пятнахвысушенной крови. Повышение уровня этого метаболита при ГА 1 происходит допоявления клинических признаков заболевания. Многие исследователи считают, чтовключение ГА1 в программы массового скрининга оправдано, так как раннее началолечения позволяет значительно улучшить качество жизни больных.
ELERS-DANLOS SYNDROME - ENZYMOPATHY, CAUSED BY HEREDITARY INSUFFICIENCY OF LYZYLOXIDASE ENZYME
A.A. Saidenova , A.A Syrykh, 2 course
Supervisor - Candidate of Biological Sciences, docent I.V. Karnaukhova
Department of Biological Chemistry
The Orenburg State Medical University
Relevance. Hereditary diseases of the connective tissue make up a rather high proportion among genetically determined diseases. One of the most common connective tissue pathologies is Ehlers-Danlos syndrome. Its frequency in the general population reaches 1: 15000 births, but the real prevalence is much higher. This is due to the fact that the disease is difficult to verify, and there are a large number of light and hidden options for the course of the disease.
Purpose.The study of the biochemical basis of Ehlers - Danlos syndrome.
Materials and methods. Review and analysis of scientific literature
Ehlers-Danlos syndrome is caused by various pathologies in the DNA regions encoding the structure of collagen, or DNA regions containing information about the biologically active proteins involved in the transformation processes of its fibers.
Currently, there are 11 types of the disease, which differ in the primary defect of the gene and the type of inheritance, the nature and severity of the clinical symptoms. Molecular mechanisms of connective tissue pathology were revealed only in certain types of the syndrome. Thus, in Ehlers-Danlos syndrome V type, lysyl oxidase deficiency is detected in fibroblast culture. Inheritance type: recessive, linked to the X chromosome.
Lysyloxidase is a copper-containing enzyme of the first class of oxidoreductases. It catalyzes the oxidation of the amino groups of some lysine residues in the tropocollagen molecule to aldehyde groups to form allisin. Next, a spontaneous aldol condensation reaction occurs between allysin and lysine, or between two allysine residues, which ensures covalent "crosslinking" of adjacent collagen molecules to each other and the formation of strong insoluble collagen fibrils.
Collagen itself, the main molecule of connective tissue, is most common in the animal world. In the tissues of mammals, there are at least five different types of collagen. In order to perform the supporting mechanical function, collagen must acquire such a structure that would be resistant to rupture. Therefore, the lack of lysyl oxidase leads to a decrease in the elasticity of the connective tissue.
The leading symptoms of this syndrome are hypermobility of the joints, moderately pronounced bleeding and significant distensibility of the skin, mitral valve is affected.
Ehlers-Danlos syndrome is not in itself fatal, and most people with a diagnosis have the opportunity to live a relatively normal life, because its symptoms are treatable. In severe cases, disability is inevitable, with the possible restriction of not only functionality, but also life expectancy.
There are no specific methods for the treatment of Ehlers-Danlos syndrome. Regular massage courses and physical therapy are important. It is recommended to conduct coursework of the main groups of drugs directly and indirectly affecting the metabolism of connective tissue.
Prevention consists in medical and genetic counseling with the determination of the degree of genetic risk of having a child with Ehlers-Danlos syndrome. In case of an identified primary biochemical defect, prenatal diagnosis is necessary. The assumption that a patient has a disease at an early clinical stage will allow them to delay potentially life-threatening situations or to ensure a high degree of preparedness for them.
Conclusion: Ehlers-Danlos syndrome remains one of the most common enzymopathies with varying degrees of severity, the specific therapy of which has not yet been developed.
ФЕНИЛКЕТОНУРИЯ И БЕРЕМЕННОСТЬ
И.А. Столяр, Н.В. Терехов, Ф.Р. Сайфутдинов, К.И. Ан, 4 курс
Научный руководитель – асс. Е.А. Логинова
Кафедра акушерства и гинекологии
Оренбургский государственный медицинский университет
PHENYLKETONURIAANDPREGNANCY
I.A. Stolyar, N.V. Terekhov, F.R. Sayfutdinov, K .I. An, 4 course
Supervisor - Ass. E.A. Loginova
Department of Biological Chemistry
The Orenburg State Medical University
Фенилкетонурияявляется врожденным и редким генетическим заболеванием, которое может нанести существенный вред для будущего ребенка. Частота данного заболевания не велика, однако, встречаются случаи, когда правильное ведение беременности у данной категории женщин может сыграть весомую роль в будущей жизни плода.
Ключевые слова:фенилкетонурия, беременность, фенилаланин.
Phenylketonuria is a congenital and rare genetic disease that can cause significant harm to an unborn baby. The frequency of this disease is not great, however, there are cases when the correct management of pregnancy in this category of women can play a significant role in the future life of the fetus.
Key words: phenylketonuria, pregnancy, phenylalanine.
Фенилкетонурия довольно таки редкое наследственное заболевание, которое связано с нарушением обмена аминокислот. Сущность состоит в том, что организм больного не может расщеплять нормально аминокислоту фенилаланин, которая относится к группе незаменимых аминокислот и поступает в организм с пищей. Из-за этого в тканях начинают накапливаться вещества, которые начинают патогенно действовать на нервную систему, в частности, на головной мозг. Из-за этого развивается умственная отсталость, вплоть до идиотии. Другое название у данной патологии – фенилпировиноградная олигофрения. Классическая фенилкетонурия (фенилкетонурия I типа) обусловлена дефицитом фермента фенилаланингидроксилаза (ФАГ), ведущим к накоплению фенилаланина и продуктов его распада в биологических жидкостях. Заболевание вызвано мутацией гена фенилаланингидроксилазы (РАН), локализующегося на длинном плече хромосомы 12, участке 12q22q24.1.
Однако, с этим заболеваниям живут и возможно нормальное функционирования организма и во взрослом состоянии. Такие пациенты могут иметь детей. Но, если родители будущего малыша знают о заболевании матери, то им необходимо принимать строгие меры к её питанию.
Главная задача акушера-гинеколога объяснить матери о важности соблюдения диеты, рассказать все риски возможные во время беременности и исхода беременности.
Клинические признаки патологии включают повышенную частоту спонтанных абортов у больных женщин, снижение массы и роста детей при рождении, умственную отсталость, микроцефалию, врожденные пороки сердца, орофациальные расщелины, экстрофию мочевого пузыря и других пороков развития.Описаны также лицевые дизморфии: гипертелоризм, широкоепереносье, вывернутые ноздри.
Основной задачей для будущей матери является ужесточение и строгое соблюдение своей диеты за 2-3 месяца до планируемой беременности. Это необходимо для снижения фенилаланина(ФА) в крови для допустимых значений для плода (2-4 мг%). Также необходимо уточнить, как будущая мать соблюдала диету в детстве и в подростковом возрасте, чтобы оценить возможность повреждения генетического аппарата её яйцеклеток.
Высокие цифры ФА в крови могут повлиять на головной мозг будущего ребенка. Если присутствует превышение порога, то существует высокая вероятность развития патологий у плода. Риск составляет примерно 90%. Причем, не обязательно то, что ребенок также будет страдать фенилкетонурией, у них могут быть только повреждения мозга и все последствия его повреждения. Этот факт был установлен в 1957 г. в ряде исследований Marbyбыли описаны подобные случаи. Так как фенилаланин активно проникает через плаценту к ребенку, то только правильно организованная диета у беременной может предотвратить данный процесс.
До зачатия уровень фенилаланина в крови должен быть снижен до 100-250 мкмоль/л (2-4 мг%) и такой уровень необходимо поддерживать в течение всей беременности. Таким образом, вероятность развития пороков сердца и других осложнений беременности в разы снижается. Согласно наблюдениям Maillot F. etal., уровень ФА в крови в течение 1-го триместра беременности контролируется у женщин 2-3 раза в неделю, во 2-м триместре - еженедельно, в 3-м - в среднем 1 раз в 2 недели; анализы крови общий и биохимический с определением уровня электролитов и витаминов проводятся 1 раз в 1,5-2 месяца, контроль тирозина в сыворотке крови – 1 раз в триместр, мониторинг прибавки массы тела - еженедельно в I-м и II-м триместрах беременности, 1 раз в две недели – в III-м триместре. Уровень фенилаланина в I и II триместрах следует поддерживать в пределах 2-4 мг/дл, в III триместре беременности – до 6 мг/дл.
В первом триместре беременности диета должна быть максимально строгой, так как этот период является критическим периодом для развития плода. Во втором и третьем триместре возможно небольшое расширение диеты беременной, но все это должно происходить под строгим контролем уровня ФА в крови и надзором врача. Необходим также прием витаминов, а также Омега-3.
Рацион будущих матерей строят согласно принципам диетического лечения классической фенилкетонурии, рекомендуемое содержание общего белка в рационе — 1,4 г/кг массы тела в сутки. Основу пищевого рациона составляют продукты растительного происхождения, приблизительно ⅔ суточной квоты белка обеспечивается за счет специализированных смесей аминокислот или гидролизатов белка.
Существуют продукты, специально предназначенные для питания женщин, планирующих беременность, и беременных, больных фенилкетонурией. К ним относятся «П-АМ универсальный», «Тетрафен 70» и отечественный продукт «Нутриген 70».
Дата добавления: 2019-07-17; просмотров: 414; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!