ГЛАВА 3. Трехкомпонентные системы. Экстракция
Теоретическая часть
Если в систему из двух компонентов, содержащую два равновесных жидких слоя, ввести небольшое количество третьего компонента, то после установления равновесия он окажется в той или иной степени присутствующим в обеих фазах. Как показывает опыт, в том случае, когда концентрация третьего компонента невелика и величина его частиц в обеих фазах одинакова, с увеличением количества третьего компонента в системе пропорционально увеличивается концентрация его в обеих фазах. Таким образом, для каждой данной температуры отношение концентрации третьего компонента в двух равновесных жидких фазах является величиной постоянной при различных его концентрациях (закон распределения) [8]. Химический потенциал зависит от активности компонента в растворе (3.1):
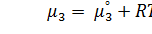 (3.1)
(3.1)
Следовательно при равновесии:
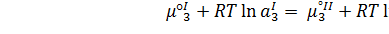 (3.2)
(3.2)
Отсюда:
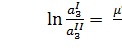 (3.3)
(3.3)
При постоянной температуре все члены в правой части уравнения (3.3) постоянны. Следовательно,
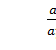 (3.4)
(3.4)
где K° - термодинамическая константа распределения. Уравнение (3.4) является выражением закона распределения Нернста-Шилова: третий компонент, добавляемый к системе, состоящей из двух взаимно нерастворимых или ограниченно растворимых жидкостей, распределяется между обоими жидкими слоями в определенном, постоянном при данной температуре отношении.
|
|
|
Сущность этого явления можно установить, рассмотрев полученные А.А. Яковкиным в 1889 г. опытные данные по распределению йода между водой и четыреххлористым углеродом. Вода и ССl4 – жидкости, не смешивающиеся друг с другом. Если в такую систему ввести небольшое количество йода и хорошо взболтать, то он будет растворяться (распределяться) как в водной, так и в другой жидкости, причем в четыреххлористом углероде растворимость йода во много раз больше, чем в воде (табл.1). Путем титрования можно определить количество йода как в той, так и в другой жидкости.
Таблица 3.1 – Распределение йода между водой и четыреххлористым углеродом
| Концентрация I2 в H2O | 0,02 | 0,04 | 0,06 | 0,08 | 0,10 |
| Концентрация I2 вCCl4 | 85,1 | 85,2 | 85,4 | 86,0 | 87,5 |
Из таблицы видно, что закон распределения действительно хорошо выполняется при малых концентрациях иода.
В основу данного исследования положен закон распределения. Если сосуществуют два раствора одного и того же компонента М в двух практически не смешивающихся растворителях А и В с общей границей раздела (т.е. соприкасающихся), то при данной температуре отношение равновесных концентраций третьего компонента М в этих фазах является постоянным и называется коэффициентом распределения (3.5):
|
|
|
КМА/В = 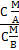 (3.5)
(3.5)
Где СМВ – концентрация компонента М в фазе В
СМА – концентрация компонента М в фазе А
КМА/В - коэффициент распределения компонента М [9].
Коэффициент распределения меняется с изменением концентрации распределяемого вещества в двух равновесных жидких фазах [10].
Закон распределения гласит: отношение концентраций вещества, распределяющегося между двумя несмешивающимися жидко- стями, является для каждой температуры величиной постоянной, не зависящей от абсолютных и относительных количеств каждого из растворителей и распределяемого вещества. Для разбавленных растворов вместо отношения активностей для расчета коэффициента распределения можно использовать отношение мольных долей растворенного вещества в обеих фазах или от- ношение концентраций:
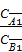 =
= 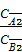 =
= 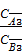 = … = К
= … = К
где СА1, СА2 , СА3… - последовательный ряд концентраций растворяемого вещества в первом растворителе;
|
|
|
СB1, СB2, СB3… – последовательный ряд концентраций растворяемого вещества во втором растворителе.
В этой форме уравнение было выведено Нернстом (в 1890 г.) и подробно исследовано Н.А. Шиловым.
Коэффициент распределения, зависящий от природы веществ и температуры, имеет линейную зависимостьК = f (CA, CB)
Приведенная формулировка верна для случая, если распределяемое вещество не изменяет при растворении своего молекулярного веса (т.е. не подвергается процессу ассоциации или диссоциации). Закон распределения не выполняется во всех случаях изменения состояния растворенных молекул хотя бы в одной из фаз системы. Такими изменениями молекул в растворе могут быть, например, диссоциация или ассоциация растворенного вещества. При этом устанавливается сложное равновесие: с одной стороны – между простыми и ассоциированными молекулами или ионами в пределах каждой фазы, с другой – между частицами, одинаковыми для всех фаз системы, распределенными между ними в некотором отношении. Тогда формула закона распределения должна быть усложнена путем введения дополнительной поправки (3.6):
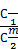 = К (3.6)
= К (3.6)
|
|
|
При достаточно полной диссоциации молекул третьего компонента на 2 или 3 частицы поправка m будет равна 2 или 3; при ассоциации двух или трех молекул – 1/2 или 1/3. Закон распределения имеет место и в случае распределения третьего компонента в двух других, находящихся в твердом агрегатном состоянии.
Закон распределения позволяет дать объяснение очень важному процессу – экстракции – и определить наиболее выгодные условия его осуществления.
Экстракция из растворов
Из закона распределения вытекает, что вещество, растворенное в одном растворителе, можно извлечь из раствора, добавляя к нему второй растворитель, не смешивающийся с первым. Такое извлечение растворенного вещества из раствора называется экстракцией [11] .
Коэффициент распределения экстрагируемого вещества можно изменять, добавляя в систему некоторые вещества, например, органические кислоты и соли органических кислот. Количественно экстракция характеризуется коэффициентом распределения (3.7):
К = 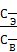 (3.7)
(3.7)
где СЭ – концентрация экстрагируемого вещества в экстрагенте;
СВ – концентрация экстрагируемого вещества в воде.
Это значит, что соотношение концентраций растворенного вещества в соприкасающихся фазах в равновесном состоянии и при данной температуре не зависит от общего его количества в смеси. К преимуществам экстракции относятся возможность разделения близкокипящих компонентов смеси, низкие рабочие температуры, относительная простота применяемой аппаратуры. В настоящее время жидкостная экстракция применяется в химической технологии, гидрометаллургии, аналитической химии для извлечения, разделения, концентрирования и очистки веществ. Экстракционные процессы используются в производстве органических продуктов, антибиотиков, пищевых продуктов, редкоземельных элементов, ряда редких, цветных и благородных металлов, в технологии ядерного горючего, при очистке сточных вод.
Материальный баланс процесса экстракции.
Если участвующие в процессе экстракции фазы практически нерастворимы, то материальный баланс процесса описывается общим уравнением. При однократном взаимодействии фаз (периодическая экстракция) материальный баланс процесса по потокам принимает вид уравнения (3.8):
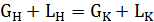 (3.8)
(3.8)
где GH, LH-количества исходного paствоpa и экстрагента соответственно, кг.
GK и LK - количества полученного экстракта и paфинатa соответственно, кг.
Уравнение может быть использовано и для непрерывного процесса при условии, что все входящие в него величины выражаются в единицах расхода, например в кг/с. Для рассматриваемого случая уравнение рабочей линии процесса экстракции описывается общим для массообменных процессов уравнением (3.9):
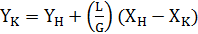 (3.9)
(3.9)
Однако чаще участвующие в жидкостной экстракции фазы обладают частичной взаимной растворимостью. Поэтому количества потоков по высоте экстрактора будут изменяться, а значит отношение L/G в уравнении не будет постоянным. Тогда очевидно, что на диаграмме у - х рабочая линия будет криволинейной. Поскольку в этом случае система является как минимум трехкомпонентной, то для анализа таких систем целесообразно воспользоваться треугольной диаграммой для построения не только равновесных, но и рабочих концентрационных зависимостей [12].
Схема процесса экстракции
Принципиальная схема экстракции приведена на рис.3.1.
В экстрактор загружаются исходный раствор F, содержащий распределяемое (экстрагируемое) вещество или вещества М, и растворитель L. Жидкость, используемая для извлечения компонентов, называется экстрагентом (Е). Массообмен между фазами протекает при их непосредственном контакте. Полученная в результате экстракции жидкая смесь поступает в разделитель, где разделяется на экстракт (Э) — раствор экстрагированных веществ в экстрагенте и рафинат (R) — остаточный раствор, из которого экстрагированы извлекаемые компоненты. Разделение смеси на экстракт и рафинат происходит в результате отстаивания или сепарирования.
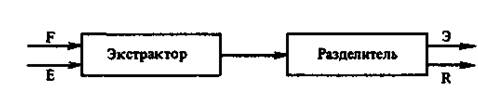
Рисунок 3.1 - Принципиальная схема экстракции
При противоточной экстракции при противоточной экстракции эффективность экстрактора измеряется эквивалентным числом ступеней. Как правило, чем больше число ступеней в экстракционной системе, тем более избирателен процесс экстракции. Однако существенной разницы между пятью и восемью ступенями при очистке смазочных масел не наблюдается.
Число ступеней экстракции, фактически обеспечиваемых в каком-либо экстракторе, зависит от режима технологического процесса, в том числе от скорости поступления сырья, вязкости исходного сырья, соотношения между растворителем и маслом, а также от температуры экстракции [13].
Практическая часть
Коэффициент распределения молочной кислоты СН3 – СН(ОН) – СООН при 250С между хлороформом и водой 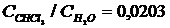 . Какое количество молочной кислоты можно извлечь из 100 см3 0,8 М молочной кислоты в хлороформе 100 см3 воды.
. Какое количество молочной кислоты можно извлечь из 100 см3 0,8 М молочной кислоты в хлороформе 100 см3 воды.
Решение:
1. Рассчитаем массу исходного компонента:
m0 = C0*V*M = 0,8*0,1*90 = 7,2 г
2. Расчитаем массу молочной кислоты после экстракции:
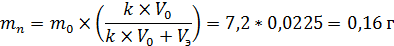
m0 - масса вещества до экстракции;
mn - масса вещества после экстракции
3. Расчитаем количество молочной кислоты:
n =  = =
= = 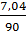 = 0,07 моль
= 0,07 моль
Таким образом, 0,07 моль молочной кислоты можно извлечь из 100 см3 0,8 М молочной кислоты в хлороформе 100 см3 воды.
ГЛАВА 4. Электрохимия
Теорeтическая часть
Гальваническим элементом называется прибор, в котором происходит превращение химической энергии в электрическую за счет окислительно-восстановительной реакции, при отсутствии непосредственного контакта между веществами и переход электронов осуществляется с помощью металлического проводника. Механизм гальванического элемента связан со структурой металла, в узлах кристаллической решетки которой находятся ионы. При погружении металла в воду, ионы, имеющиеся на поверхности, гидратируются полярными молекулами воды и переходят из пластинки в раствор, оставляя на пластинке электроны, которые заряжают ее отрицательно. Вследствие электростатического притяжения, ионы цинка из раствора притягиваются к цинковой пластинке, что препятствует дальнейшему переходу ионов цинка в раствор, устанавливается подвижное равновесие и образуется двойной электрический слой (ДЭС). Скачок потенциала, возникающий на границе между металлом и раствором, называется электродным потенциалом. Чем активнее металл, тем больше ионов переходит в раствор и тем больше величина отрицательного заряда. Так как цинковая пластинка заряжается отрицательно, то такой электродный потенциал считается отрицательным.
На медном электроде происходит иное явление: энергия электронно-ионной связи в медной пластинке больше, чем в цинке, поэтому катионы меди переходят из раствора на пластинку в большем количестве, чем с поверхности металла в раствор, и медная пластинка заражается менее отрицательно, чем цинковая, а прилегающий к ней слой жидкости отрицательно. Такой электродный потенциал считается положительным. Электродом гальванического элемента называется система, состоящая из металла, погруженного в раствор ионов этого же металла. При соединении медной и цинковой пластинок металлическим проводником ионы более активного металла (цинка) переходят в раствор, адсорбируются на пластинке и электроны, имеющиеся в избытке на цинковой пластинке пойдут от цинковой пластинки к медной, в результате чего возникнет электрический ток. Электроны, попадая на медную пластинку, нейтрализуют ионы меди, имеющиеся в растворе и ионы меди осаждаются на медной пластинке. Уменьшение электронов на цинковой пластинке компенсируется переходом в раствор новых ионов цинка, нарушается ДЭС, при этом сульфат-ионы переходят от медной пластинке к цинковой. Электрод, на котором идет процесс окисления (отдача электронов), называется анодом. Электрод, на котором идет процесс восстановления (присоединения электронов), называется катодом [14].
Для определения ЭДС этого элемента нужно сравнить стандартные электродные потенциалы обоих электродов. При записи электродных реакций принято, что окисленная форма находится в левой части, а восстановленная – в правой части уравнения (4.1 – 4.2).
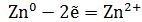 (4.1)
(4.1)
 (4.2)
(4.2)
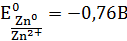
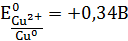
где E0 – электродвижущая сила (ЭДС) гальванического элемента, когда все реагенты в стандартном состоянии. ЭДС элемента вычисляется вычитанием из потенциала катода потенциала анода.
ЭДС элемента равна +0,34 – (–0,76) = 1,1В; чем больше электродные потенциалы отличаются друг от друга, тем больше ЭДС. Если погрузить металл в раствор соли большей концентрации, то потенциал нестандартный. Значит, на величину электродного потенциала влияет концентрация и температура. Такая зависимость выражается уравнением В. Нернста (4.3):
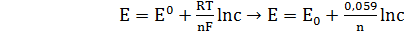 (4.3)
(4.3)
где n – число ионов;
R – универсальная газовая постоянная;
Т – температура;
С – концентрация активных ионов в растворе;
F – число Фарадея = 96500В.
Нернст предложил считать условным нулём потенциал водородного электрода при концентрации водородных ионов в растворе, равной 1, и давлении газообразного водорода 1 атм. Эта условная шкала потенциалов называется водородной шкалой. В настоящее время применяется главным образом условная водородная шкала, в которой при всех температурах за ноль выбран потенциал стандартного водородного электрода. Она отличается от первоначальной водородной шкалы Нернста тем, что в ней вместо единичных концентраций и давления выбраны единичная активность и летучесть. Это условие позволяет определять потенциалы электродов в водородной шкале при любых Т, однако при каждой Т потенциал водородного электрода может быть иным, то есть условный нуль не будет одним и тем же при разных Т.
Таким образом, электродным потенциалом электрода называется ЭДС элемента, составленного из этого электрода (справа) и стандартного водородного электрода (слева), например (4.4):
(+) Pt ïH2 ç H+, aq çç Zn2+ ç Zn (-) (4.4)
ЭДС этого элемента (ЕZn2+çZn) отрицательна (– 0,763 В при активности ионов цинка в растворе, равной 1; это и есть стандартный электродный потенциал цинка). Чтобы найти электродный потенциал меди, нужно составить элемент (4.5)
(-) Pt ïH2 ç H+, aq çç Cu2+ ç Cu (+) (4,5)
Здесь ЭДС цепи (ЕCu2+çCu) положительна (+ 0,337 В при активности ионов меди, равной 1, - стандартный электродный потенциал меди).
Целесообразно в схеме полуэлемента записывать сочетание электрод + раствор иона в том порядке, который имеется в записи элемента, составленного из стандартного водородного электрода и данного; именно для записанного таким образом электрода следует приводить электродный потенциал с соответствующим знаком. При обратной записи следует изменить знак потенциала, например:
(1) Zn2+, aq çZn ; Е = - 0,763 B , (2) Zn çZn2+, aq ; Е = + 0,763 B .
Только первый тип записи приводит к тем знакам величин Е, которые соответствуют электродным потенциалам. Величины, соответствующие записи (2), не следует называть электродными потенциалами, но ими можно пользоваться при подсчете ЭДС цепи (для электродов, расположение которых в схеме цепи является обратным расположению их в сочетании с водородным электродом), например (4.6):
(-) Zn çZn2+, aq çCu2+, aq ç Cu (+) (4,6)
Диффузионный потенциал, возникающий на границе растворов ZnSO4 - CuSO4, усложняет расчет.
Электроды первого рода
К электродам первого рода относятся электроды, состоящие из металлической пластинки, погруженной в раствор соли того же металла. При обратимой работе элемента, в который включен электрод, на металлической пластинке идет процесс перехода катионов из металла в раствор либо из раствора в металл. Таким образом, электроды первого рода обратимы по катиону и их потенциал связан уравнением Нернста с концентрацией катиона. К электродам первого рода относят также и водородный электрод.
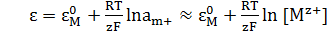 (4.7)
(4.7)
Электродами второго рода являются электроды, в которых металл покрыт малорастворимой солыо этого металла и находится в растворе, содержащем другую растворимую соль с тем же анионом. Электроды этого типа обратимы относительно аниона, и зависимость их электродного потенциала от температуры и концентрации аниона может быть записана в следующем виде (4.8):
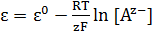 (4.8)
(4.8)
Электроды сравнения предназначены для измерения электродных потенциалов. Необходимость их использования обусловлена невозможностью измерения абсолютной величины потенциала отдельного электрода. В качестве электрода сравнения может быть использован электрод, обладающий постоянным и не зависящим от состава раствора потенциалом. При этом необязательно знать числовую величину потенциала. Значение потенциала должно воспроизводиться ине изменяться от опыта к опыту. Существенными требованиями к электродам сравнения являются низкое электрическое сопротивление, отсутствие влияния на состав анализируемого раствора, способность не вызывать появления значительного диффузионного потенциала и, несомненно, простота конструкции.
Универсальным электродом сравнения является стандартный водородный электрод, но для практической работы он неудобен из-за необходимости использования очень чистого водорода и ряда других причин.
Насыщенный каломельный электрод (НКЭ) схематически изображен на рисунке 4.1. Это стеклянный сосуд с впаянным в него солевым мостиком, который погружают в анализируемый раствор. На дно сосуда наливается чистая ртуть, в которую погружена платиновая проволока (контакт), впаянная в стеклянную трубку. Другой конец платиновой проволоки припаивается к медному проводнику, идущему к клемме потенциометра. Поверх ртути помещается паста каломели (Hg2Cl2), тщательно растертая со ртутью и хлористым калием. Для заполнения каломельного электрода используют насыщенный раствор хлорида калия (КСlнас.). Систему насыщенного каломельного электрода обозначают Pt|Hg|Hg2Cl2|KClнас. В тех случаях, когда попадание ионов хлора в анализируемый раствор нежелательно, каломельный электрод можно использовать с дополнительным солевым мостиком, заполненным другим электролитом.
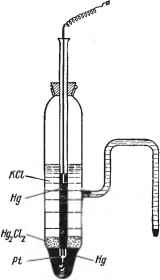
Рисунок – 4.1 Насыщенный каломельный электрод
Потенциал каломельного электрода определяется реакцией 2Hg + 2Cl- ↔ Hg2Cl2тв. + 2e, и его изменение соответствует при E0 = 0,244 уравнению Нернста (4.9)
E = E0 – 0.058*lg [Cl-] (4.9)
При этом концентрации веществ, находящихся в твердой фазе, постоянны. Если раствор является насыщенным по хлориду калия, то его потенциал не зависит от концентрации ионов хлора в растворе, находящихся в избытке. Тогда каломельный электрод используют как электрод сравнения.
Хлорсеребряный электрод (ХСЭ) состоит из серебряной проволоки с наплавленным слоем хлорида серебра, погруженной в насыщенный раствор хлорида калия - Ag|AgClтв|КClнас.|Cl-. Потенциал такого электрода определяется равновесной системой Ag++ Cl- ↔ AgClтв + e и рассчитывается по уравнению (4.10)
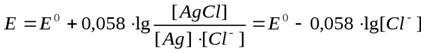 (4.10)
(4.10)
Следовательно, потенциал хлорсеребряного электрода (Е) при избыточной концентрации [Cl-] (KClнас) при 25 0С равен Е0 (+ 0,222 В).
Чаще всего в качестве электрода сравнения используется насыщенный хлорсеребряный электрод, потенциал которого зависит только от температуры. В отличие от каломельного, он устойчив при повышенных температурах и применим как в водных, так и во многих неводных средах.
Окислительно-восстановительные электроды.
В отличие от описанных электродных процессов в случае окислительно-восстановительных электродов процессы получения и отдачи электронов атомами или ионами происходят не на поверхности электрода, а только в растворе электролита. Если опустить платиновый (или другой инертный) электрод в раствор, содержащий двух- и трехзарядные ионы железа и соединить этот электрод проводником с другим электродом, то возможно либо восстановление ионов Fe3+ до Fe2+ за счет электронов, полученных от платины, либо окисление ионов Fe2+ до Fe3+ с передачей электронов платине. Сама платина в электродном процессе не участвуют, являясь лишь переносчиком электронов. Такой электрод, состоящий из инертного проводника первого рода, помещенного в раствор электролита, содержащего один элемент в различных степенях окисления, называется окислительно-восстановительным или редокс-электродом. Потенциал окислительно-восстановительного электрода также определяют относительно стандартного водородного электрода.
Зависимость потенциала редокс-электрода ɛRO от концентрации (активности) окисленной [Ox] и восстановленной форм [Red] для окислительно-восстановительной реакции, в которой не участвуют никакие другие частицы, кроме окислителя и восстановителя, имеет следующий вид (здесь n – число электронов, участвующих в элементарном акте окислительно-восстановительной реакции)(4.11):
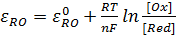 (4.11)
(4.11)
В случае более сложных систем в выражении для окислительно-восстановительного потенциала фигурируют концентрации всех участвующих в реакции соединений, т.е. под окисленной формой следует понимать все соединения в левой части уравнения реакции (4.12):
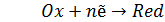 (4.12)
(4.12)
а под восстановленной – все соединения в правой части уравнения. Так, для окислительно-восстановительных реакций, протекающих с участием ионов водорода (4.13):
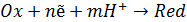 (4.13)
(4.13)
уравнение Нернста будет записываться следующим образом (4.14):
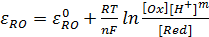 (4.14)
(4.14)
При составлении гальванических элементов с участием редокс-электрода электродная реакции на последнем в зависимости от природы второго электрода может быть либо окислительной, либо восстановительной. Например, если составить гальванический элемент из электрода Pt / Fe3+, Fe2+ и второго электрода, имеющего более положительный электродный потенциал, то при работе элемента редокс-электрод будет являться анодом, т.е. на нем будет протекать процесс окисления (4.15):
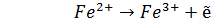 (4.15)
(4.15)
Если потенциал второго электрода будет меньше, чем потенциал электрода Pt / Fe3+, Fe2+, то на последнем будет протекать реакция восстановления и он будет являться катодом (4.16):
 (4.16)
(4.16)
Знание величин электродных потенциалов позволяет определить возможность и направление самопроизвольного протекания любой окислительно-восстановительной реакции при одновременном наличии в растворе двух или более окислительно-восстановительных пар. Восстановленная форма любого элемента или иона будет восстанавливать окисленную форму другого элемента или иона, имеющего более положительный электродный потенциал.
Практическая часть
Задача: Для окислительно-восстановительного элемента типа
Pt | 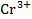 ,
, 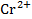 ||
|| 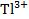 ,
, 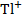 | Pt
| Pt
по стандартным электродным потенциалам полуэлементов напишите уравнение и вычислите константы равновесия реакции окисления – восстановления. Вычислите ЭДС элемента при 298 К. Примите, что активности aН2О =1, аН2 = 0,1.
Решение:
+Pt | | , || , | Pt-
Вычислим значения стандартных электродных потенциалов полуэлементов:
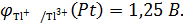
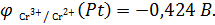
На электродах протекают реакции:
(A) + 2ē →
(B) 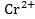 - ē →
- ē →
Суммарная реакция, протекающая при работе электрохимического элемента, имеет вид:
+ 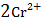 → +
→ + 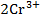
Вычислим значения потенциалов для каждого электрода:
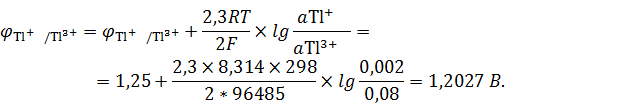
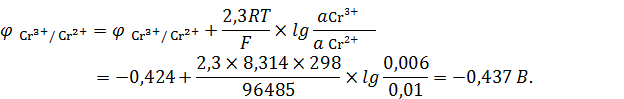
Подставив числовые значения, получим:
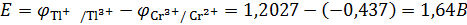
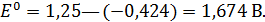
По уравнению определим константу равновесия реакции, протекающей в гальваническом элементе:
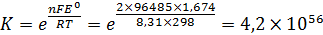
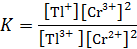
ГЛАВА 5 Химическая кинетика
Теоретическая часть
Ско́рость хими́ческой реа́кции — изменение количества одного из реагирующих веществ за единицу времени в единице реакционного пространства. Является ключевым понятием химической кинетики. Скорость химической реакции — величина всегда положительная, поэтому, если она определяется по исходному веществу (концентрация которого убывает в процессе реакции), то полученное значение умножается на −1.
Например, для реакции: А + В → С + D выражение для скорости выглядит: 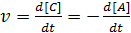
В 1865 году Н. Н. Бекетовым и в 1867 году Гульдбергом и Вааге был сформулирован закон действующих масс: скорость химической реакции в каждый момент времени пропорциональна концентрациям реагентов, возведенным в степени, равные их стехиометрическим коэффициентам.
Для элементарных реакций показатель степени при значении концентрации каждого вещества часто равен его стехиометрическому коэффициенту, для сложных реакций это правило не соблюдается. Кроме концентрации на скорость химической реакции оказывают влияние следующие факторы:
1) природа реагирующих веществ,
2) наличие катализатора,
3) температура (правило Вант-Гоффа, Уравнение Аррениуса),
4) давление(P).
5) площадь поверхности реагирующих веществ.
Если мы рассмотрим самую простую химическую реакцию A + B → C, то мы заметим, что мгновенная скорость химической реакции величина непостоянная.
Изучение различных кинетических кривых показывает, что скорость уменьшения концентрации реагента со временем падает. Наблюдаемое падение скорости, очевидно, связано с уменьшением концентрации реагентов. Кинетические исследования подтверждают правильность этого предположения, выражаемого в наиболее общем виде с помощью закона действующих масс для скорости реакции. Закон действующих масс не применяется для твёрдых веществ.
Реакции первого порядка
Рассмотрим зависимость от времени концентрации исходного вещества А для случая реакции первого порядка А→В. Реакции первого порядка характеризуются кинетическим уравнением вида (5.1-5.2):
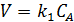 (5.1)
(5.1)
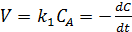 (5.2)
(5.2)
После интегрирования выражения получаем (5.3):
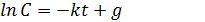 (5.3)
(5.3)
Константу интегрирования g определим из начальных условий: в момент времени t = 0 концентрация С равна начальной концентрации С0. Отсюда следует, что g = ln С0. Получаем (5.4):
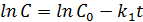 (5.4)
(5.4)
Т.о., логарифм концентрации для реакции первого порядка линейно зависит от времени и константа скорости численно равна тангенсу угла наклона прямой к оси времени.
 (5.6)
(5.6)
Из уравнения легко получить выражение для константы скорости односторонней реакции первого порядка (5.7):
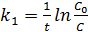 (5.7)
(5.7)
Еще одной кинетической характеристикой реакции является период полупревращения t1/2 – время, за которое концентрация исходного вещества уменьшается вдвое по сравнению с исходной. Выразим t1/2 для реакции первого порядка, учитывая, что С = ½С0 :
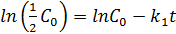 (5.8)
(5.8)
Отсюда
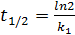 (5.9)
(5.9)
Как видно из полученного выражения, период полупревращения реакции первого порядка не зависит от начальной концентрации исходного вещества.
Реакции второго порядка
Для реакций второго порядка кинетическое уравнение имеет следующий вид (5.10):
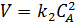 (5.10)
(5.10)
Либо (5.11):
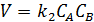 (5.11)
(5.11)
Рассмотрим простейший случай, когда кинетическое уравнение имеет вид 1 или, что то же самое, в уравнении вида 2 концентрации исходных веществ одинаковы; уравнение 1 в этом случае можно переписать следующим образом (5.12):
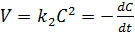 (5.12)
(5.12)
После разделения переменных и интегрирования получаем (5.13):
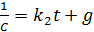 (5.13)
(5.13)
Постоянную интегрирования g, как и в предыдущем случае, определим из начальных условий. Получим (5.14):
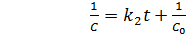 (5.14)
(5.14)
Т.о., для реакций второго порядка, имеющих кинетическое уравнение вида 1, характерна линейная зависимость обратной концентрации от времени и константа скорости равна тангенсу угла наклона прямой к оси времени (5.16):
 (5.16)
(5.16)
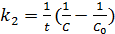 (5.17)
(5.17)
Если начальные концентрации реагирующих веществ C0,А и C0,В различны, то константу скорости реакции находят интегрированием уравнения в котором CА и CВ – концентрации реагирующих веществ в момент времени t от начала реакции (5.18):
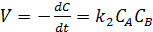 (5.18)
(5.18)
В этом случае для константы скорости получаем выражение (5.19)
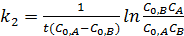 (5.19)
(5.19)
Порядок химической реакции есть формально-кинетическое понятие, физический смысл которого для элементарных (одностадийных) реакций заключается в следующем: порядок реакции равен числу одновременно изменяющихся концентраций. В случае элементарных реакций порядок реакции может быть равен сумме коэффициентов в стехиометрическом уравнении реакции; однако в общем случае порядок реакции определяется только из экспериментальных данных и зависит от условий проведения реакции. Рассмотрим в качестве примера элементарную реакцию гидролиза этилового эфира уксусной кислоты (этилацетата), кинетика которой изучается в лабораторном практикуме по физической химии (5.20):
СН3СООС2Н5 + Н2О → СН3СООН + С2Н5ОН (5.20)
Если проводить эту реакцию при близких концентрациях этилацетата и воды, то общий порядок реакции равен двум и кинетическое уравнение имеет следующий вид (5.21):
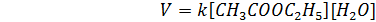 (5.21)
(5.21)
При проведении этой же реакции в условиях большого избытка одного из реагентов (воды или этилацетата) концентрация вещества, находящегося в избытке, практически не изменяется и может быть включена в константу скорости; кинетическое уравнение для двух возможных случаев принимает следующий вид:
1) Избыток воды:
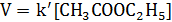
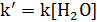
2) Избыток этилацетата:
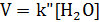
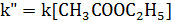
В этих случаях мы имеем дело с т.н. реакцией псевдопервого порядка. Проведение реакции при большом избытке одного из исходных веществ используется для определения частных порядков реакции.
Влияние температуры на количество столкновений молекул может быть показано с помощью модели. В первом приближении влияние температуры на скорость реакций определяется правилом Вант-Гоффа (сформулировано Я. Х. Вант-Гоффом на основании экспериментального изучения множества реакций):
В интервале температур от 0oС до 100oС при повышении температуры на каждые 10 градусов скорость химической реакции возрастает в 2-4 раза(5.22):
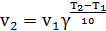 (5.22)
(5.22)
где 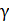 - температурный коэффициент, принимающий значения от 2 до 4.
- температурный коэффициент, принимающий значения от 2 до 4.
Объяснение зависимости скорости реакции от температуры было дано С.Аррениусом. К реакции приводит не каждое столкновение молекул реагентов, а только наиболее сильные столкновения. Лишь молекулы, обладающие избытком кинетической энергии, способны к химической реакции [16].
С.Аррениус рассчитал долю активных (т.е. приводящих к реакции) соударений реагирующих частиц a, зависящую от температуры и вывел уравнение Аррениуса для константы скорости реакции (5.23):
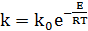 (5.23)
(5.23)
где k0 и E зависят от природы реагентов.
Е - это минимальная энергия, которой должны обладать реагирующие частицы, чтобы столкновение между ними привело к реакции. Энергию активации выражают в джоулях на моль (Дж/моль).
В случае простых реакций величина ЕА показывает, какой минимальной (избыточной по сравнению со средней) энергией в расчете на 1 моль должны обладать реагирующие частицы, чтобы они могли вступить в химическую реакцию. В случае сложных реакций величина ЕА называется эмпирической или кажущейся энергией активации и в общем случае сложным образом зависит от энергий активации отдельных стадий данной реакции.
За величину энергии активации приближенно принимают превышение средней энергии активированного комплекса над средним уровнем энергии исходных веществ. Она зависит от природы реагирующих веществ и характеризует изменение скорости реакции от температуры. Чем больше энергия активации, тем быстрее увеличивается с ростом температуры скорость реакции [16].
Энергию активации можно определить зная константы скорости лишь при двух температурах (5.24):
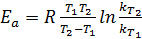 (5.24)
(5.24)
Рассмотрим путь некоторой элементарной реакции: А+В -> С Поскольку химическое взаимодействие частиц связано с разрывом старых химических связей и образованием новых, считается, что всякая элементарная реакция проходит через образование некоторого неустойчивого промежуточного соединения, называемого активированным комплексом
Образование активированного комплекса всегда требует затраты некоторого количества энергии, что вызвано, во-первых, отталкиванием электронных оболочек и атомных ядер при сближении частиц и, во-вторых, необходимостью построения определенной пространственной конфигурации атомов в активированном комплексе и перераспределения электронной плотности. Таким образом, по пути из начального состояния в конечное система должна преодолеть своего рода энергетический барьер. Энергия активации реакции приближённо равна превышению средней энергии активированного комплекса над средним уровнем энергии реагентов. Очевидно, что если прямая реакция является экзотермической, то энергия активации обратной реакции Е'A выше, нежели энергия активации прямой реакции ЕA. Энергии активации прямой и обратной реакции связаны друг с другом через изменение внутренней энергии в ходе реакции. Вышесказанное можно проиллюстрировать с помощью энергетической диаграммы химической реакции (рис. 5.1).
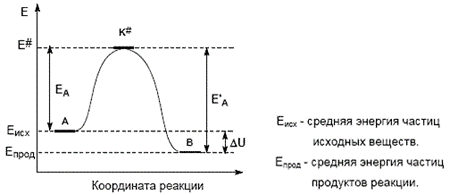
Рисунок 5.1 - Энергетическая диаграмма химической реакции
Практическая часть
Задача: По значениям констант скоростей реакции при двух температурах определите энергию активации, константу скорости при температуре Т3, температурный коэффициент скорости. Составить кинетическое уравнение, учесть, что порядок реакции и молекуклярность совпадают. Рассчитать количество вещества, израсходованное за время t, если начальные концентрации равны С0.
Таблица 5.1
| Реакция | T1, K | k1 | T2, K | k2 | T3, K | t, мин | C0, моль/л |
| 2NO2→2N2+О2 | 986 | 6,72 | 1165 | 977 | 1053 | 65 | 1,75 |
Решение:
1. Найдем температурный коэффициент скорости:
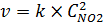
n = 2
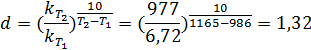
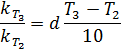
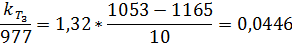

2. Найдем энергию активации:
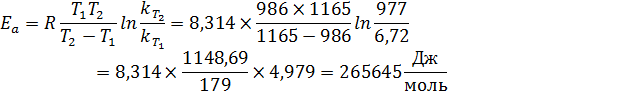
3. Найдем константу скорости при T3:
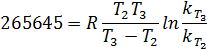
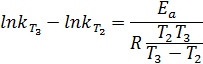
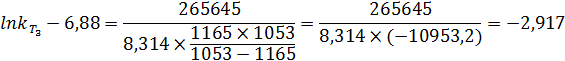
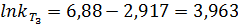
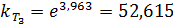
4. Рассчитаем количество вещества, израсходованного за время t, при начальной концентрации C0.
V=k∙C2
k∙t = 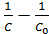
C = 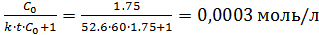
x = C0 – C
x = 1.75 – 0,0003 = 1.749 моль/л
ЗАКЛЮЧЕНИЕ
К главным задачам физической химии можно отнести изучение и объяснение основных закономерностей, определяющих направленность химических процессов, скорость их протекания, влияние на них среды, примесей, излучения и т.п., условия получения максимального выхода продуктов.
При решении основных задач физическая химия пользуется следующими методами физики:
1) Термодинамический метод – применяется для исследования направленности процессов, законов химических и фазовых равновесий.
2) Метод статистической физики – дает возможность рассчитать свойства макроскопических тел, исходя из свойств частиц, образующих эти тела.
3) Квантово-механический метод – устанавливает способ описания и законы движения микрочастиц, а также связь величин, характеризующих частицы и системы, с физическими величинами, непосредственно измеряемыми на опыте.
С одной стороны физическая химия является экспериментальной наукой: с использованием экспериментальных физических и химических методов исследования строения вещества, структуры молекул, элементарных актов химических взаимодействий (рентгенография, оптические методы, радио- и масс-спектроскопия и др.). С другой стороны – наука теоретическая, т. к. имеет свой богатый математический аппарат. Позволяет теоретически рассчитать строение молекул, возможность их взаимодействия друг с другом, скорость реакции, ее тепловой эффект и др. Тесное сочетание теории и эксперимента в физико-химических исследованиях позволяет успешнее решать научные и прикладные задачи в химии и в сложных областях науки и техники.
Дата добавления: 2018-08-06; просмотров: 554; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!