ГЛАВА 2. Фазовое равновесие в жидких бинарных системах, образованных двумя летучими компонентами
МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ РОССИЙСКОЙ ФЕДЕРАЦИИ
СТЕРЛИТАМАКСКИЙ ФИЛИАЛ
ФЕДЕРАЛЬНОГО ГОСУДАРСТВЕННОГО БЮДЖЕТНОГО
ОБРАЗОВАТЕЛЬНОГО УЧРЕЖДЕНИЯ
ВЫСШЕГО ОБРАЗОВАНИЯ
«БАШКИРСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ»
Естественнонаучный факультет
Кафедра химии и химической технологии
КУРСОВАЯ РАБОТА
ПРИЛОЖЕНИЯ ХИМИЧЕСКОЙ ТЕРМОДИНАМИКИ. КИНЕТИКА
| Работа допущена к защите Заведующий кафедрой химии и химической технологии д.т.н., профессор ________ /Абдрашитов Я.М./ «___» ____________2018 г. Научный руководитель: к.т.н, доцент _________ /Опарина Ф.Р./ «___» ____________2018 г. | Выполнил студент 3 курса группы ЕНФ-ХИМ31 очного отделения Галимова Зухра Исмагиловна ___________ Работа защищена «___» ____________2018 г. Оценка _______________ |
СОДЕРЖАНИЕ
ВВЕДЕНИЕ. 3
ГЛАВА 1. Основы химической термодинамики. Равновесие. 4
1.1 Теоретическая часть. 4
1.2 Практическая часть. 10
ГЛАВА 2. Фазовое равновесие в жидких бинарных системах, образованных двумя летучими компонентами. 12
2.1 Теоретическая часть. 12
2.2 Практическая часть. 22
ГЛАВА 3. Трехкомпонентные системы. Экстракция. 25
3.2 Теоретическая часть. 25
3.2 Практическая часть. 31
ГЛАВА 4. Электрохимия. 32
4.1 Теорeтическая часть. 32
4.2 Практическая часть. 40
ГЛАВА 5 Химическая кинетика. 42
5.1 Теоретическая часть. 42
5.2 Практическая часть. 49
ЗАКЛЮЧЕНИЕ. 51
СПИСОК ИСПОЛЬЗОВАННЫХ ИСТОЧНИКОВ.. 52
|
|
|
ВВЕДЕНИЕ
Физическая химия изучает законы, управляющие химическими процессами, связь этих процессов со свойствами участвовших в них веществ и условиями, при которых они протекают. Благодаря этому появляется возможность управления химическими процессами и отыскания оптимальных условий их проведения.
Устанавливая общие законы физико-химических процессов, физическая химия является теоретическим обобщением неорганической, органической и аналитической химии.
Физическая химия включает в себя несколько разделов, характеризующих направление развития этой науки и определяющих ее предмет. Это в первую очередь химическая термодинамика с ее тремя основными законами, фазовым и химическим равновесиями и учение о растворах и адсорбции веществ. Сюда же относят и термодинамику неравновесных процессов, которая, по существу, является границей раздела между термодинамикой и кинетикой.
Главная особенность современной физической химии - широкое применение разнообразных физических методов экспериментального исследования, стремление выяснить детальный молекулярный механизм химических реакций. Физическая химия даёт теоретические основы для исследований как в областях неорганической, органической и аналитической химии, так и в разработке химической технологии.
|
|
|
ГЛАВА 1. Основы химической термодинамики. Равновесие
Теоретическая часть
Термохи́мия — раздел химической термодинамики, в задачу которой входит определение и изучение тепловых эффектов реакций, а также установление их взаимосвязей с различными физико-химическими параметрами. Ещё одной из задач термохимии является измерение теплоёмкостей веществ и установление их теплот фазовых переходов.
В ходе реакции происходит разрыв связей в исходных веществах и образование новых связей в продуктах реакции. Поскольку образование связи идет с выделением, а ее разрыв - с поглощением энергии, то химические реакции сопровождаются энергетическими эффектами. Энергия выделяется, если рвущиеся связи в исходных веществах менее прочны, чем связи, образующиеся в продуктах реакции, в противном случае - энергия поглощается. Обычно энергия выделяется и поглощается в форме теплоты, т.е. химическая форма энергии преобразуется в тепловую. Таким образом, химические реакции сопровождаются тепловыми эффектами.
Тепловой эффект - количество теплоты, выделившееся или поглощенное химической системой при протекании в ней химической реакции [1].
|
|
|
Тепловой эффект обозначается символами Q или ∆H (Q = -∆H). Его величина соответствует разности между энергиями исходного и конечного состояний реакции:
∆H = Hкон.- Hисх. = Eкон.- Eисх.
Реакции, протекающие с выделением теплоты, проявляют положительный тепловой эффект (Q>0, ∆H<0) и называются экзотермическими.
Реакции, которые идут с поглощением теплоты из окружающей среды (Q<0, ∆H>0), т.е. с отрицательным тепловым эффектом, являются эндотермическими.
Закон Гесса — основной закон термохимии, который формулируется следующим образом:
Тепловой эффект химической реакции, проводимой в изобарно-изотермических или изохорно-изотермических условиях, зависит только от вида и состояния исходных веществ и продуктов реакции и не зависит от пути её протекания.
Иными словами, количество теплоты, выделяющееся или поглощающееся при каком-либо процессе, всегда одно и то же, независимо от того, протекает ли данное химическое превращение в одну или в несколько стадий (при условии, что температура, давление и агрегатные состояния веществ одинаковы).
Закон Гесса является математическим следствием первого закона термодинамики. Он составляет теоретическую основу термохимии - раздела физической химии, занимающегося изучением тепловых эффектов различных химических процессов. В термохимии при написании химического уравнения принято включать в него тепловой эффект реакции ΔН и агрегатное состояние участников реакции. Такие уравнения называют термохимическими. Поскольку ΔН зависит от термодинамических параметров, систему приводят к стандартному состоянию при р=1 атм и Т = 298 К, а стандартную энтальпию реакции обозначают ΔН0298. Большое практическое значения имеют следствия из закона Гесса:
|
|
|
а) изменение энтальпии химической реакции не зависит от числа ее промежуточных стадий, т.е. суммарный тепловой эффект реакции на каком-либо одном пути равен суммарному тепловому эффекту на любом другом пути;
б) энтальпии прямой химической реакции равна взятой с противоположным знаком энтальпии обратной химической реакции;
в) тепловой эффект химической реакции Δ0Hr равен разности между суммой теплот образования продуктов реакции (конечных веществ) и сумм теплот образования исходных (начальных) веществ, взятых с соответствующими стехиометрическими коэффициентами.
д) тепловой эффект реакции равен разности сумм теплот сгорания исходных веществ и сумм теплот сгорания продуктов реакции, взятых с соответствующими стехиометрическими коэффициентами [2]..
Таким образом, пользуясь табличными значениями теплот образования или сгорания веществ, можно рассчитать теплоту реакции, не прибегая к эксперименту. Табличные величины теплот образования и сгорания веществ обычно относятся к т. н. стандартным условиям. Для расчёта теплоты процесса, протекающего при иных условиях, необходимо использовать и другие законы термохимии, например, закон Кирхгофа, описывающий зависимость теплового эффекта реакции от температуры
Теплоемкость системы, соответствующая бесконечно малому изменению температуры, называется истинной теплоемкостью (1.1):
с =  =
=  (1.1)
(1.1)
Когда теплоемкость относится к единице массы (1кг, 1г), то она называется удельной теплоемкостью и измеряется в [кДж/(кг.К)] или [Дж/(г .К)]; когда она относится к одному моль вещества - молярной тепло- емкостью с размерностью[Дж/(моль.К)]. Молярная теплоемкость связана с удельной соотношением с = суд*М, где М - молярная масса, кг/кмоль или г/моль.
Если начальное и конечное состояния химической реакции (реакций) совпадают, то её (их) тепловой эффект равен нулю.
S – функция состояния системы, называемая энтропией. Энтропия характеризует меру неупорядоченности (хаотичности) состояния системы. Единицами измерения энтропии являются Дж/(моль•К).
При абсолютном нуле температур (Т = 0 К) энтропия идеального кристалла любого чистого простого вещества или соединения равна нулю. Равенство нулю S при 0 К позволяет вычислить абсолютные величины энтропий веществ на основе экспериментальных данных о температурной зависимости теплоемкости.
Изменение энтропии в процессе выражается уравнением (1.2):
∆S =∑ S(прод.) – ∑S(исх.) (1.2)
где S(прод.) и S(исх.) – соответственно абсолютные энтропии продуктов реакции и исходных веществ.
На качественном уровне знак ∆S реакции можно оценить по изменению объема системы ∆V в результате процесса. Знак ∆V определяется по изменению количества вещества газообразных реагентов ∆nг. Так, для реакции CaCO3(к) = CaO(к) + CO2(г):
(∆nг = 1) ∆V > 0, значит,∆ S > 0.
Для реакции С(графит) + 2Н2(г) = СН4(г)
(∆nг = -1) ∆V < 0, следовательно и ∆S < 0.
Величины энтропии принято относить к стандартному состоянию. Чаще всего значения S рассматриваются при Р = 101,325 кПа (1 атм) и температуре Т = 298,15 К (25ºС). Энтропия в этом случае обозначается Sо298 и называется стандартной энтропией при Т = 298,15 К. Следует подчеркнуть, что энтропия вещества S (Sº) увеличивается при повышении температуры.
Стандартная энтропия образования Sºf,298 (или∆ Sºобр,298) – это изменение энтропии в процессе образования данного вещества (обычно 1 моль), находящегося в стандартном состоянии, из простых веществ, также находящихся в стандартном состоянии.
Физический смысл энтропии может быть пояснен следующим мысленным экспериментом. Пусть идеальный кристалл какого-либо вещества, например хлорида натрия, охлажден до абсолютного нуля температуры. В этих условиях ионы натрия и хлора, составляющие кристалл, становятся практически неподвижными, и данное макроскопическое состояние характеризуется одним единственным микросостоянием, т.е. W=1, и в соответствии с (3.13) S=0. При повышении температуры ионы начнут колебаться около положений равновесия в кристаллической решетке, число микросостояний, соответствующих одному макросостоянию, возрастает, и, следовательно, S>0. Таким образом, энтропия является мерой неупорядоченности состояния системы. Энтропия системы увеличивается во всех процессах, сопровождающихся уменьшением упорядоченности (нагревание, растворение, испарение, реакции разложения и т.п.). Процессы, идущие с увеличением упорядоченности (охлаждение, кристаллизация, сжатие и т.п.), приводят к уменьшению энтропии.
Энтропия является функцией состояния, но в отличие от большинства других термодинамических функций возможно экспериментальное определение абсолютного значения энтропии вещества. Эта возможность основана на постулате М. Планка, согласно которому при абсолютном нуле энтропия идеального кристалла равна нулю(третий закон термодинамики)[3].
Стандартной теплотой образования (энтальпией образования) вещества называется энтальпия реакции образования 1 моля этого вещества из элементов (простых веществ, то есть состоящих из атомов одного вида), находящихся в наиболее устойчивом стандартном состоянии. Стандартные энтальпии образования веществ ∆H0 298 (кДж/моль) приводятся в справочниках. При использовании справочных значений необходимо обращать внимание на фазовое состояние веществ, участвующих в реакции. Энтальпия образования наиболее устойчивых простых веществ равна 0.
Энергия Гиббса
G – функция состояния системы, называемая энергией Гиббса. Энергия Гиббса равна (1.3):
ΔG = ΔН – ТΔS (1.3)
Абсолютное значение энергии Гиббса определить невозможно, однако можно вычислить изменение G в результате протекания процесса.
Критерий самопроизвольного протекания процесса: в системах, находящихся при Р, Т = const, самопроизвольно могут протекать только процессы, сопровождающиеся уменьшением энергии Гиббса (∆G < 0). При достижении равновесия в системе ∆G = 0.
Стандартная энергия Гиббса образования ∆ Gºf,298 (или образования ∆ Gºобр,298) – это изменение энергии Гиббса в процессе образования данного вещества (обычно 1 моль), находящегося в стандартном состоянии, из простых веществ, также находящихся в стандартном состоянии, причем простые вещества присутствуют в наиболее термодинамически устойчивых состояниях при данной температуре.
Для простых веществ, находящихся в термодинамически наиболее устойчивой форме образования ∆ Gºf,298 = 0.
Практическая часть
Задача: При температуре Т, К и давлении Р=1 атм рассчитать: изменение энтальпии  , изменение энтропии
, изменение энтропии  , изменение энергии Гиббса
, изменение энергии Гиббса  и константу равновесия Кр. Объяснить в какую сторону сместится равновесие реакции:
и константу равновесия Кр. Объяснить в какую сторону сместится равновесие реакции:
а) при повышении температуры;
б) при повышении давления.
CH2ClCH2Cl(г) = CH2CHCl(г) + HCl(г) T = 800 K
Решение:
1. Вычислим изменение энтальпии ∆H.
Найдем стандартную энтальпию реакции:
∆H298 = [∆Hf(CH2CHCl) + ∆Hf(HCl)] ‒ [ ∆Hf(CH2ClCH2Cl)] = (35,15+(-92,31)-(-129,70) = 72,54 кДж/моль ‒ реакция эндотермическая, т.к. ∆H ˃ 0, с поглощением теплоты.
По закону Кирхгогофа:
∆HT = ∆H298 + 
Найдем разность изобарных теплоемкостей продуктов реакции и исходных веществ:
Cp = [Cp(CH2CHCl)] + [Cp(HCl) - Cp(CH2ClCH2Cl)] = 53,72+29,14 – 78,66 = 4,2 Дж/моль∙К
∆H800 = 72,54 +  = 74,6484 кДж/моль
= 74,6484 кДж/моль
2. Вычислим изменение энтропии ∆S.
Найдем стандартную энтропию реакции:
∆S298 = [∆S(CH2CHCl) + ∆S(HCl)] - [∆S(CH2ClCH2Cl)] = (263,93+186,79) - 308,19 = 142,53 Дж/моль∙К
∆ST = ∆S298 + ∆Cp ln 
∆S800 = 142,53 + 4,2 ∙ ln  = 146,67 Дж/моль∙К
= 146,67 Дж/моль∙К
3. Вычислим изменение энергии Гиббса ∆G.
∆GT = ∆HT ‒ T∆ST
∆G800 = (74,6484 ∙ 103 ) ‒ 800 ∙ 146,67 = ‒42687,6 Дж/моль∙К
∆G < 0, (G → Gmin, энергия Гиббса убывает), то процесс в данном направлении термодинамически возможен
4. Вычислим константу равновесия Кр.
ln Кр = ‒ 
ln Кр = ‒  = 6,5
= 6,5
Кр = e6,5
Кр = 665,14
Кр ‒ константа равновесия, выраженная через парциальные давления газов.
P(CH2CHCl) + P(HCl) + P(CH2ClCH2Cl) = 1 атм
P(HCl) ∙ P(CH2CHCl)
Кр = ——————
P(CH2ClCH2Cl)
При повышении температуры равновесие химической реакции смещается вправо.
При повышении давления равновесие химической реакции смещается влево.
ГЛАВА 2. Фазовое равновесие в жидких бинарных системах, образованных двумя летучими компонентами
Теоретическая часть
Азеотропная смесь – смесь двух или более жидкостей, состав которой не меняется при кипении, то есть смесь с равенством составов равновесных жидкой и паровой фаз.
Фазой называется часть гетерогенной системы, ограниченная четко выраженной поверхностью раздела, характеризующаяся одинаковыми свойствами во всех точках при условии отсутствия сил внешних физических полей.
По числу фаз системы делятся на однофазные, двухфазные трехфазные и многофазные. Индивидуальные вещества, которые могут быть выделены из системы и существовать самостоятельно вне ее, называются составляющими веществами системы [4].
Вещество, которое может быть выделено из системы и существовать в не ее, называется компонентом.
Компоненты являются независимыми составляющими веществами системы. При химическом взаимодействии в системе число компонентов находят как разность между числом составляющих веществ системы и числом уравнений, связывающих между собой концентрации этих веществ. В противном случае число компонентов равно числу составляющих веществ системы. Различают однокомпонентные, двухкомпонентные и многокомпонентные системы.
Условия фазового равновесия. Фазовым гетерогенным равновесием называется равновесие в системе, состоящей из двух или большего числа фаз.
К фазовым равновесиям относятся равновесия типа:
Т1 ® Т2, Т ® Ж, Т ® Г, Ж1 ® Ж2, Ж ® Г,
где Т, Ж, Г обозначают соответственно твердое тело, жидкость, газ.
Условия равновесия гетерогенной системы можно записать в следующем виде:
T1 = T2 = ... = TФ,
P1 = P2= ... = PФ,
mi1 = mi2 = ... = miФ.
где верхние индексы относятся к фазам, а нижние – к компонентам.
Анализ уравнений для термодинамической системы, состоящей из Ф фаз, каждая из которых состоит из K компонентов, дает следующее соотношение (2.1):
C = К – Ф + 2, (2.1)
где C – число термодинамических степеней свободы.
Уравнение (2.1) называется правилом фаз Гиббса:в равновесной термодинамической системе, на которую из внешних факторов оказывают влияние только температура и давление, число термодинамических степеней свободы равно числу компонентов минус число фаз плюс два.
Правило фаз – это соотношение между числом независимых компонентов, числом фаз и числом степеней свободы при равновесии.
Число степеней свободы возрастает с увеличением числа компонентов и уменьшается с увеличением числа фаз. Поскольку число степеней свободы не может быть отрицательным, то число фаз в равновесной системе не может превышать величину K + 2.
Если на систему действует электрическое или магнитное поля, поле тяготения и другие физические поля в сумме, равной n различных факторов, то число термодинамических степеней свободы записывается в виде (2.2)
C = K – Ф + n (2.2)
Если из внешних факторов на систему оказывает влияние только температура (при P = const) или только давление (при Т = const), то число термодинамических степеней свободы выражается уравнением (2.3)
Cусл = K – Ф + 1 (2.3)
Эти системы называются условно инвариантными. При постоянстве температуры и давления число термодинамических степеней свободы определяется выражением (2.4)
Cусл = K – Ф (2.4)
Если составы двух равновесных фаз, например, жидкости и пара, одинаковы, то при подсчете числа степеней свободы следует учитывать уравнение, связывающее концентрации компонентов Сж = Сп, и тогда (2.5)
Cусл = K – Ф + 2 – 1 = К – Ф + 1 (2.5)
Для однокомпонентной системы, на равновесие которой из внешних факторов оказывают влияние только температура и давление, правило фаз Гиббса выражается формулой (2.6)
C = 3 – Ф (2.6)
Другими словами, в однокомпонентной системе число фаз, находящихся в равновесии, не может быть больше трех.
Из правила фаз следует, что для однокомпонентной системы максимальное число фаз определяется из условия (2.7):
Ф = К – С + 1 = 1 – 0 + 2 = 3 (2.7)
Для двухкомпонентной системы (2.8):
Ф = К – С + 1 = 2 – 0 + 2 = 4 (2.8)
Правилу фаз Гиббса не подчиняются коллоидные системы, в которых дисперсная фаза микроскопических размеров (фазы должны обладать макроскопическими размерами и поверхностную энергию можно не учитывать). Правило фаз неприменимо к неравновесным метастабильным состояниям (переохлажденная вода, перегретый пар и т.д.).
Жидкие бинарные смеси представляют собой растворы двух жидких летучих компонентов. Летучими называются жидкости, имеющие при комнатной температуре высокое давление пара (относительно низкую температуру кипения). Согласно классификации растворы подразделяют на идеальные, предельно разбавленные и реальные. В идеальных растворах взаимодействия между молекулами одного вида и молекулами разных видов одинаковы и их смешение не приводит к изменению объема ΔVсм=0 и не сопровождается тепловым эффектом ΔHсм=0. Если компоненты раствора имеют молекулы близкого состава с одинаковой полярностью, или содержание одного из компонентов настолько мало, что взаимодействиями между молекулами разного вида можно пренебречь, то свойства такого раствора близки к свойствам идеального раствора, такой раствор называют предельно разбавленным [5].
Идеальные растворы подчиняются закону Рауля (2.9):
Парциальное давление насыщенного пара компонента раствора прямо пропорционально его мольной доле в растворе, причем коэффициент пропорциональности равен давлению насыщенного пара над чистым компонентом.
Pi = Pi˚ Xi (2.9)
Парциальным давлением компонента в газовой смеси называется давление, которое бы он создавал, если бы занимал весь объем смеси.
Согласно закону Дальтонадавление в любой газовой смеси, в том числе, давление пара над раствором летучих компонентов равно сумме парциальных давлений компонентов (2.10)
p = p1 + p2 (2.10)
и, соответственно (2.11)
p = p1˚∙X1 + p2˚∙X2 (2.11)
Таким образом, зависимость давления пара над идеальным раствором двух жидких летучих компонентов от состава раствора имеет вид прямой линии. Для изучения фазовых равновесий жидкость – пар используют диаграммы состояния или фазовые диаграммы, представляющие собой зависимости состав – свойство. График зависимости давления пара от состава при постоянной температуре приведен на рисунке 2.1.
Верхняя кривая (abc) показывает зависимость давления пара p от состава раствора, нижняя (adc) – зависимость давления пара p от состава пара. В области, расположенной выше кривой abc, устойчива жидкая фаза. Область, заключенная между этими кривыми, является гетерогенной: пар находится в равновесии с жидкостью.

Рисунок 2.1. – Диаграмма состояния
Смесь, фигуративная точка которой К находится в этой области, распадается на две равновесные фазы – жидкую состава 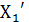 и пар состава
и пар состава  .
.
Первый закон Рауля нетрудно вывести, используя понятие химического потенциала компонента, если считать пар над раствором идеальным газом. Условием гетерогенного равновесия является равенство химических потенциалов компонентов системы во всех фазах. Химический потенциал растворителя в растворе μА(р) связан с мольной долей растворителя в растворе XА уравнением (2.12):
ΜА(р) = μ˚А(р) + RTln XA (2.12)
Химический потенциал растворителя в паре μА(п) можно выразить через парциальное давление пара растворителя РА (2.13):
μА(п) = μ˚А(п) + RTln PA (2.13)
Химический потенциал чистого жидкого растворителя μ*А равен химическому потенциалу равновесного пара (2.14):
Μ*А = μ˚А(п) + RTln P˚A (2.14)
В состоянии равновесия μА(п) = μА(р). Комбинируя выражения, легко получить (2.15):
ΜА(р) ‒ μ*А = RTln  (2.15)
(2.15)
Принимая, что μ*А = μ°А(р), получаем следующее уравнение (2.16):
RTln XA = RTln  (2.16)
(2.16)
Отсюда легко получить выражение для первого закона Рауля (2.19):
Ln XA = ln (2.17)
XA =  (2.18)
(2.18)
PA = PA˚XA (2.19)
Таким образом, для идеальных бинарных растворов зависимость общего и парциального давления насыщенного пара от состава раствора, выраженного в мольных долях компонента В, является линейной при любых концентрациях (рис. 2.2). К таким системам относятся, например, системы бензол – толуол, гексан – гептан, смеси других изомерных углеводородов. Для реальных растворов данные зависимости являются криволинейными (рис. 2.3) [6].

Рисунок 2.2 - Зависимость парциальных и общего давлений пара идеального раствора от концентрации

Рисунок 2.3 - Зависимость парциальных и общего давлений пара идеальных и реальных бинарных растворов от состава при положительных (слева) и отрицательных (справа) отклонениях от закона Рауля.
Таким образом, зависимость давления пара над идеальным раствором двух жидких летучих компонентов от состава раствора имеет вид прямой линии. В случае реальных растворов межмолекулярные взаимодействия могут приводить как к положительным, так и к отрицательным отклонениям от закона Рауля и, соответственно, отклонению уравнения р=f(x)от линейной зависимости. Экспериментально установлено, что состав пара над раствором в общем случае не совпадает с составом раствора, находящегося в равновесии с паром.
Относительное понижение давления насыщенного пара растворителя над раствором равно мольной доле растворенного вещества (2.20).
 = N2, (2.20)
= N2, (2.20)
где
p0 - давление насыщенного пара над чистым растворителем;
p - давление насыщенного пара растворителя над раствором;
N2 - мольная доля растворенного вещества;
n1 - число молей растворенного вещества;
n2 - число молей растворителя.
Понижение давления насыщенного пара растворителя над раствором нелетучего вещества приводит к повышению температуры кипения и понижению температуры замерзания раствора по сравнению с чистым растворителем, что находит отражение на диаграмме состояния растворителя
Следствия закона Рауля
1. Понижение температуры замерзания DТзам и повышение температуры кипения DТкип раствора неэлектролита прямо пропорциональны количеству вещества, растворенному в данном количестве растворителя.
2. Эквимолярные (т.е. содержащие одно и то же число молей) количества растворенных веществ, будучи растворены в одном и том же количестве данного растворителя, одинаково понижают температуру его замерзания и одинаково повышают температуру его кипения.
Выделить из смеси один из компонентов системы возможно ректификацией. Ректификация — разделение жидких смесей на практически чистые компоненты, отличающиеся температурами кипения, путём многократных испарений жидкости и конденсации паров.
Ректификация позволяет достичь высокой степени разделения смесей на составляющие их компоненты. Процесс осуществляется путем многократного контакта между неравновесными жидкой и паровой фазами, движущимися противотоком. При взаимодействии происходит массообмен, обусловленный стремлением системы к состоянию равновесия. В результате каждого контакта компоненты смеси перераспределяются между фазами так, что пара обогащается НКК, а жидкость – ВКК. Многократное контактирование в итоге приводит в итоге к практически полному разделению смеси [7].
Процесс осуществляют периодически или непрерывно (наиболее часто в нефтепереработке) при атмосферном или повышенном давлении, а также под вакуумом. Повышенное давление используют для разделения смесей компонентов с низкими температурами кипения, а вакуум – для разделения смесей высококипящих веществ, чтобы избежать их разложения при высоких температурах. Взаимодействие паров и жидкости достигается в ректификационных колоннах, снабженных контактными устройствами - ректификационными тарелками или насадкой.
Сырье, которое необходимо разделить на две части - высококипящую и низкокипящую, подается в среднюю часть колонны на тарелку питания. Сырье может подаваться в колонну в виде жидкости, пара пли парожидкостной смеси.
Введенная в колонну жидкая смесь стекает по контактным устройствам в нижнюю часть колонны, называемую отпарной. Навстречу потоку жидкости поднимаются пары, образующиеся в результате кипения жидкости в кубе колонны. Пары, поступающие на тарелку с нижележащей, имеют более высокую температуру, чем стекающая с вышележащей тарелки жидкость. На тарелке в результате контакта паров и жидкости (флегмы) происходит выравнивание температур. При этом из паров, которые охлаждаются, выделяется в жидкую фазу некоторое количество высококипящего компонента, а из стекающей жидкости испаряется некоторое количество низкокипящего компонента, т. е. на каждой тарелке или контактном устройстве происходит теплообмен и массообмен. В парах по мере их подъема по колонне уменьшается содержание в.к.к. и соответственно возрастает концентрация н.к.к., а в опускающейся флегме возрастает концентрация в.к.к. и уменьшается концентрация н.к.к.
Пары с верха колонны отводятся в конденсатор, где они охлаждаются, частично или полностью конденсируются. Часть сконденсированного верхнего продукта или дистиллята закачивается насосом в качестве орошения, которое, стекая с верхней тарелки, создает жидкостный поток — флегму. Избыточная часть дистиллята откачивается за пределы установки или же направляется в качестве сырья для другой колонны.
Флегма с низа колонны отводится в кипятильник, где она в результате подвода теплоты подвергается частичному испарению. Выделившиеся из флегмы пары из кипятильника возвращаются в колонну (под нижнюю тарелку) и образуют восходящий паровой поток, что необходимо для ректификации.
Практическая часть
Задача: дана зависимость составов состава жидкой фазы и находящегося с ней в равновесии пара от температуры для двухкомпонентной жидкой системы А – В при постоянном давлении. Молярный состав жидкой фазы х и насыщенного пара у выражен в процентах вещества А. По приведенным данным:
1. Построить график зависимости состава пара (Х) от состава жидкой фазы (У) при постоянном давлении;
2. Построить диаграмму кипения системы А-В;
3. Проанализируйте фазовое состояние системы; укажите области жидкой (Ж), паровой (П) и гетерогенной (Ж-П) систем;
4. Укажите состав азеотропного раствора;
5. Определить температуру кипения системы с мольной долей а% вещества А. Укажите состав первого пузырька пара над этой системой; температуру, при которой закончится кипение системы; каков состав последней капли жидкой фазы?
6. Определить состав пара, находящегося в равновесии с жидкой фазой, кипящей при температуре Т1;
7. Рассчитать количество компонента, который может быть выделен ректификацией из системы, состоящей из b кг. вещества А и с кг. вещества В.
Таблица 2.1 – Зависимость: состав-температура
| Система | Р×10-4, Па | Молярная доля А, % | Т, К | |
| х – жидкая фаза | у - пар | |||
| А-CS2 В-CН3CОСН3 | 10,133 | 0,0 | 0,0 | 329,2 |
| 1,9 | 8,3 | 327,0 | ||
| 4,8 | 18,5 | 324,4 | ||
| 13,4 | 35,1 | 319,6 | ||
| 18,6 | 44,3 | 317,0 | ||
| 29,1 | 52,8 | 314,4 | ||
| 38,0 | 57,4 | 313,3 | ||
|
|
| 44,8 | 59,8 | 312,8 |
| 53,6 | 62,7 | 312,3 | ||
| 65,3 | 66,1 | 312,1 | ||
| 78,9 | 70,5 | 312,3 | ||
| 87,9 | 76,0 | 313,5 | ||
| 96,8 | 88,6 | 316,5 | ||
| 100,0 | 100,0 | 319,3 | ||
Таблица 2.2 – Температура-состав
| T1, K | a, % моль | b, кг | с, кг |
| 317 | 25 | 30,4 | 69,6 |
Решение:
1. Построили график зависимости состава пара от состава жидкости. См. График 1.
2. Построили диаграмму кипения системы А-В. См. График 2.
3. На графике указали области жидкой, паровой и гетерогенной систем.
4. Состав азеотропного раствора: А=67%, B=33%.
5. Для определения температуры кипения системы необходимо провести перпендикуляр от оси Х, соответствующий содержанию в системе 25% CS2. Пересечение перпендикуляра с кривой состава кипящей жидкости (нижней кривой диаграммы) показывает точку начала кипения системы, соответствующей 315,4 К. Для определения состава паровой фазы необходимо провести изотерму через точку начала кипения. Пересечение изотермы с кривой насыщенного пара показывает состав пара 49% CS2 и 51% CН3CОСН3. (Это будет состав первого пузырька пара над системой). Состав последней капли жидкой фазы - 8% CS2 и 92% CН3CОСН3.
6. Проводим изотерму Т=317, ее пересечение с кривой состава паровой фазы дает две точки . По ним определяем состав парообразной фазы. Соответственно, 44% CS2 и 67 % CН3CОСН3 (слева от азеотропной точки) .
7. Рассчитаем какой компонент и в каком количестве может быть выделен из системы, состоящей из 30,4 кг CS2 (компонент А) и 69,6 кг CН3CОСН3 (компонент В). Найдем мольную долю компонента А.


∑𝑛=0,4+1,2=1,6 кмоль


Состав азеотропной смеси:
Компонент А 0,4 – 67%.
Компонент В Х – 33%.

m(В) = 0,197*58 = 11,426 кг
Найдем чистый компонент В:
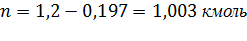
m В = 1,003*58 = 58,174кг
Азеотропная смесь: (30,4 кг А + 11,426 кг В)
Чистый компонент В: (58,174 кг)
Итого: m(A)+m(B)+m(Аз) = 30,4+58,174+11,426 = 100кг
Фазовая диаграмма позволяет определить состав продуктов ректификации и составить материальный баланс.
Дата добавления: 2018-08-06; просмотров: 618; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!