Ретроспективний аналіз розвитку оптичних методів аналізу
У 1960 р. святкувалося сторіччя з дня відкриття Кірхгофом і Бунзеном спектрального аналізу. Це відкриття провело глибоке враження на сучасників і мало величезне значення для розвитку всієї системи наших знань про навколишній світ. Досить сказати, що без спектрального аналізу ми до цих пір нічого не знали б про склад небесних тіл, якщо не говорити про метеорити, що зрідка потрапляють до наших рук.
Незадовго до великого відкриття Кірхгофа і Бунзена французький філософ О. Конт писав, що у людства немає ніякої надії дізнатися, з чого складаються Сонце і зірки. Пройшло декілька років, і спектроскоп Кірхгофа спростував цей песимістичний прогноз, як було спростовано багато інших прогнозів філософів і теологів, що намагалися обмежити можливості людського пізнання на підставі загальних положень своїх учень і релігій [1].
Перші п'ятдесят років спектрального аналізу були роками його найбільших успіхів. Після того, як було встановлено, що кожному хімічному елементу належить свій спектр, що є такою ж характерною ознакою елементу, як дактилоскопічний відбиток − ознакою людини, почалася гонитва за спектрами. Виявилось, що слабкі промені світла, тисячі років, що йдуть до нас, від віддалених зірок і туманностей, доносять величезну кількість відомостей про світи, які вони покинули. Дослідження спектрального складу цих променів дає відомості не тольки про склад, але також про температуру і про рух зірки.
З перших днів свого існування спектральний аналіз допоміг зробити ряд важливих відкриттів. Направивши спектроскоп на Сонці в 1861 р., Кірхгоф проводить гігантську роботу, що трохи не привела його до повної сліпоти: він складає перший атлас сонячного спектру і порівнює його із спектрами ряду елементів. З неспростовною переконливістю доводить він присутність в хромосфері заліза і висловлює достатньо обгрунтоване припущення про існування в хромосфері елементів: Ca, Mg, Na, Ni, Cr. Присутність в хромосфері З, Ba, Cu і Zn розглядається ним як вірогідне. Можна тільки дивуватися з об'єму отриманих результатів, переконливості доказів і ретельності вимірювань, які були досягнуті Кірхгофом при грубій апаратурі і обмежених експериментальних можливостях того часу (рисунок 1.1).
Так вперше були отримані свідчення про склад Сонця. Згодом досліджувалися також спектри зірок і туманностей. Не менш тріумфальними були спостереження спектрів земних об'єктів.
У 1861 р. Кірхгоф і Бунзен відкривають два нові елементи – лужні метали цезій и рубідій. У тому ж році Крукс, досліджуючи спектр відходів виробництва сірчаної кислоти, відкриває талій. У 1863 р. Райх і Ріхтер виявляють яскраві сині лінії в спектрі одного зразка цинкової обманки і відкривають індій.

Рисунок 1.1 – Спектроскоп Кірхгофа і Бунзена
У 1875 р. Лекок де Буабодран за спектром цинкової обманки з Піренеїв виявляє новий елемент, споріднений індію, − галій. У 1868 р. англійський астроном Локьєр виявив яскраву жовту лінію в спектрі хромосфери. Він приписав її новому елементу, названому ним гелієм. Локьєр досить легко придумував гіпотези, які потім не підтверджувалися, але цій гіпотезі повезло: у 1875 р. Рамзай виділив інертний газ з мінералу клеєвіта і точними вимірюваннями довів тотожність випромінюваної ним лінії з лінією, приписаною гелію. Так гелій з гіпотетичної сонячної речовини перетворився на повноправного члена періодичної системи елементів. Це був один з найбільших тріумфів спектрального аналізу.
В кінці XIX століття за допомогою спектроскопа Рамзай і Релей відкривають аргон, і незабаром Рамзай і Траверс знаходять і решту інертних газів: неон, ксенон і криптон. В кінці XIX і початку XX століття Демарсе, Лекок де Буабодран і Урбен досліджують спектри рідкісних земель. Тільки завдяки спектроскопії вдається встановити 14 індивідуальних елементів цієї групи. Додамо сюди ще відкритий в 1923 р. гафній — останній елемент, який був виявлений по його спектру (правда, вже рентгенівському). Разом спектроскопії належить заслуга відкриття 25 елементів. Це приблизно 30% всіх елементів, що існують в земній корі. У цій цифрі, мабуть, найпереконливіше виявляється значення спектрального методу. З самого початку його розвитку стало ясно, що спектроскопія є дуже чутливим методом − з її допомогою можна відкривати такі кількості елементу, які недоступні для звичайного хімічного аналізу [1].
Відкриттям ряду нових елементів в основному була завершена перша блискуча епоха розвитку спектрального аналізу. Не все, звичайно, протікало гладко. Так, наприклад, спектроскоп відкрив в деяких туманностях новий елемент, названий «небулієм», а в спектрі сонячної корони були спостережені лінії, приписані елементу «коронію». Згодом небулій виявився киснем, а короній − кальцієм. На початок першої світової війни спектральний аналіз, як дуже чутливий і швидкий спосіб хімічного аналізу, застосовувався для вирішення багатьох завдань, труднодоступних для інших методів. З його допомогою, наприклад, француз де Грамон довів, що вибухи на вулицях Парижа походять від снарядів «Великої Берти», з якої німці обстрілювали місто з небаченої раніше дистанції (120 км.), а майбутній чарівник оптичного експерименту Роберт Вуд ще в 1891 р. наочно продемонстрував, що в студентській їдальні одного з американських університетів студентів годують печенею, приготованою із залишків недоїдених вчора біфштексів. Не дивлячись на такий широкий діапазон проблем, доступних спектральному аналізу, він поки не використовувався широко в аналітичній практиці. Причин цьому було дві. По-перше, до 20-х років спектральний аналіз був чисто якісним, в кращому разі напівкількісним методом. З його допомогою можна було дізнатися, чи присутній елемент, що цікавить нас, в пробі; можна було відповісти на питання, багато або мало цього елементу, але зміряти його зміст з будь-який точністю не вдавалося. Такий метод аналізу не годився для більшості технічних завдань. Друга причина полягала в рідкості і дорожнечі апаратури, а також у малій кількості людей що володіли спектрально-аналітичною методикою: для хіміків вона була дуже складна і незвичайна, фізики стояли далеко від аналітичних завдань.
У першій чверті двадцятого століття було в основному закінчено створення якісного спектрального аналізу: вивчені спектри більшості елементів, і складені таблиці цих спектрів, встановлені найбільш придатні для аналізу лінії, добре розроблена техніка фотографування і вимірювання спектрів. До кінця цього періоду почалася розробка методів кількісного спектрального аналізу і додаток цих методів до вирішення ряду виробничих завдань.
У Радянському Союзі Г. С. Ландсберг і Д. С. Рождественський на початку 30-х років організували лабораторії, завданням яких був розвиток і впровадження в промисловість методів спектрального аналізу. На той час вже стало ясно, що спектроскопія може успішно конкурувати з іншими хімічними методами аналізу, а у ряді випадків володіє серйозними перевагами перед ними. Розробка методів спектрального аналізу пішла по шляху підвищення його точності, чутливості і продуктивності. Крім того, конструювалася апаратура і долалася недовіра до нового методу з боку прихильників класичної аналітичної школи. Останнє, здається, було найважчим етапом, оскільки ще і сьогодні можна почути думку, що хоча спектральний аналіз і дуже чутливий, але його точність дуже мала [1]. Все ж таки зараз вже ніхто не сумнівається в тому, що спектральний аналіз належить до основних методів дослідження складу речовини, і приблизно з 30-х років до теперішнього часу відбувається безперервне вдосконалення методів якісного і кількісного спектрального аналізу і все більш широке його проникнення в техніку для вирішення чисто практичних завдань контролю виробництва металів і реактивів, геологічної розвідки і ряду інших.
До оптичних методів аналізу відносять фізико-хімічні методи, засновані на взаємодії електромагнітного випромінювання з речовиною. Ця взаємодія приводить до різних енергетичних переходів, які реєструються експериментально у вигляді поглинання випромінювання, віддзеркалення і розсіяння електромагнітного випромінювання. Оптичні методи включають велику групу спектральних методів аналізу.
У методах атомної спектроскопії ми маємо справу з вузькими лінійчатими спектрами, а в методах молекулярної спектроскопії – з широкими слабоструктурованими спектрами. Це визначає можливість їх застосування в кількісному аналізі і вимоги, що пред'являються до вимірювальної апаратури – спектральних приладів.
Фотометричний метод
Метод аналізу, заснований на переведенні визначуваного компоненту в з'єднання з подальшим визначенням кількості цього компоненту, що поглинає світло, шляхом вимірювання світлопоглинання розчину отриманої сполуки, називається фотометричним.
За забарвленням розчинів забарвлених речовин можна визначати концентрацію того або іншого компоненту або візуально, або за допомогою фотоелементів − приладів, що перетворюють світлову енергію на електричну. Відповідно до цього розрізняють фотометричний візуальний метод аналізу, званий часто колориметричним, і метод аналізу із застосуванням фотоелементів − власне фотометричний метод аналізу. Фотометричний метод є об'єктивним методом, оскільки результати його не залежать від здібностей спостерігача, на відміну від результатів колориметричного − суб'єктивного методу [1].
Фотометричний метод аналізу − один з найстаріших і поширеніших методів фізико-хімічного аналізу. Його розповсюдженню сприяли порівняльна простота необхідного устаткування, особливо для візуальних методів, висока чутливість і можливість застосування для визначення майже всіх елементів періодичної системи і великої кількості органічних речовин. Відкриття все нових і нових реагентів, які створюють забарвлені сполуки з неорганічними іонами і органічними речовинами, робить в даний час застосування цього методу майже необмеженим.
Фотометричний метод аналізу може застосовуватися для великого діапазону визначуваних концентрацій. Його використовують як для визначення основних компонентів різних складних технічних об'єктів із змістом до 20 − 30% визначуваного компоненту, так і для визначення мікродомішок в цих об'єктах при змісті їх до 10-3 − 10-4%. Комбінування фотометричних методів з деякими методами розділення − хроматографічним, екстракційним дозволяє на 1−2 порядки підвищити чутливість визначення, довівши його до 10-5.
В деяких випадках фотометричний метод може бути застосований для одночасного визначення в розчині декількох іонів, хоча, як це буде показано нижче, його можливості обмежені.
Дуже цінне використання фотометричних методів для вирішення багатьох теоретичних питань аналітичної і фізичної хімії.
Здатність хімічної сполуки, неорганічного іона і органічного угрупування поглинати променисту енергію певних довжин хвиль використовується у фотометричному аналізі.
Серед неорганічних речовин порівняно небагато сполук, що володіють власним забарвленням; це сполуки марганцю (VII), хрому (VI), мідь (II) і деякі інші. У ряді випадків забарвлені сполуки утворюються при взаємодії неорганічних реагентів, наприклад, виникнення яскраво-червоного забарвлення при взаємодії заліза (III) з роданідом, нікелю (II) з аміаком і деякі інші, проте і таких реакцій порівняно мало. Зазвичай для колориметричних визначень неорганічних іонів доводиться використовувати численні реакції їх з органічними реактивами, що супроводжуються утворенням забарвлених сполук [1].
Слід зазначити, що утворення забарвлених у видимій області спектру з'єднань необхідне тільки для візуальних методів колориметричного аналізу. При визначенні інструментальними методами можуть бути використані лінії і смуги поглинання, які лежать як в ультрафіолетовій, так і в інфрачервоних областях спектру.
В даний час майже для всіх неорганічних іонів є досить великі набори органічних реактивів, що взаємодіють з утворенням забарвлених з'єднань.
Характер забарвлення залежить також від ряду інших чинників про що буде сказано нижче.
Таким чином, кожна речовина володіє здатністю поглинати променисту енергію у вигляді квантів енергії, відповідних певним довжинам хвиль. Лінії або смуги поглинання розташовуються в ультрафіолетовій, видимій або інфрачервоній областях спектру. Ці смуги і лінії можуть бути використані для якісного і кількісного фотометричного аналізу [1].
Основний закон фотометрії. Якщо світловий потік інтенсивності 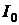 падає на кювету, що містить досліджуваний розчин, то частина цього потоку
падає на кювету, що містить досліджуваний розчин, то частина цього потоку  відбивається від стінок кювети і поверхні розчину, частина його
відбивається від стінок кювети і поверхні розчину, частина його 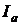 поглинається молекулами речовини, що міститься в розчині, і витрачається на зміну електронній, обертальній і коливальній енергії цих молекул, частина енергії
поглинається молекулами речовини, що міститься в розчині, і витрачається на зміну електронній, обертальній і коливальній енергії цих молекул, частина енергії 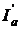 поглинається молекулами самого розчинника. Якщо в розчині присутні тверді частинки у вигляді мутей або суспензій, то частина світлової енергії
поглинається молекулами самого розчинника. Якщо в розчині присутні тверді частинки у вигляді мутей або суспензій, то частина світлової енергії  відбивається і від цих частинок і, нарешті, частина енергії
відбивається і від цих частинок і, нарешті, частина енергії 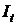 проходить через кювету. На підставі закону збереження енергії можна записати рівняння:
проходить через кювету. На підставі закону збереження енергії можна записати рівняння:
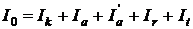 . .
| (1.1) |
При аналізі прозорих розчинів в рівнянні (1.1) член дорівнює 0. При роботі впродовж всього дослідження з одним розчинником член можна вважати постійним. Крім того розчинники завжди підбирають так, щоб вони самі в досліджуваній області спектру мали мінімальне поглинання яким можна нехтувати. При використанні однієї і тієї ж кювети значення відбитого світлового потоку дуже мало і ним можна нехтувати. Тому приведене вище рівняння (1.1) можна спростити:
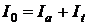 . .
| (1.2) |
Безпосередніми вимірюваннями можна визначити інтенсивність падаючого світлового потоку ( ), що пройшов через аналізований розчин ( ). Значення може бути знайдене за різницею між та ; безпосередньому ж вимірюванню ця величина не піддається.
На підставі численних експериментів П. Бугером, а потім і І. Ламбертом був сформульований закон, що встановлює, що шари речовин однакової товщини, за рівних умов, завжди поглинають одну і ту ж частину падаючого на них світлового потоку. Якщо припустити, що при проходженні через шар даної товщини інтенсивність світлового потоку зменшується в два рази, можна побудувати графічну залежність інтенсивності світлового потоку від товщини шару (рисунок 1.2).
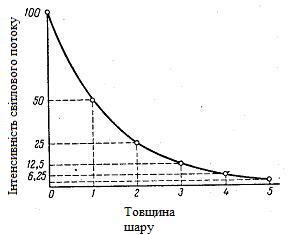
Рисунок 1.2 – Крива залежності інтенсивності світлового потоку, який пройшов від товщини поглинаючого шару
Математично ця залежність виражається рівнянням:
 , ,
| (1.3) |
де − інтенсивність світлового потоку після проходження шару;
− інтенсивність падаючого світлового потоку;
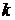 − коефіцієнт поглинання, що характеризує поглинання світла даним тілом і залежить від властивостей даного тіла;
− коефіцієнт поглинання, що характеризує поглинання світла даним тілом і залежить від властивостей даного тіла;
 – товщина шару.
– товщина шару.
З даного закону випливає:
1) відношення інтенсивності світлового потоку, що пройшов через шар розчину, до інтенсивності падаючого світлового потоку не залежить від абсолютної інтенсивності падаючого світлового потоку;
2) якщо товщина шару розчину збільшується за арифметичною прогресією, інтенсивність світлового потоку, що пройшов через нього, зменшується за геометричною прогресією.
Поглинальна здатність будь-якого розчину може бути цілком охарактеризована значенням коефіцієнта . Коефіцієнт поглинання залежить лише від природи розчиненої речовини і довжини хвилі падаючого світла. Отже, закон поглинання світла Бугера-Ламберта справедливий тільки для монохроматичного світла, тобто для світла певної довжини хвилі.
Вивчаючи поглинання світла розчинами, Бер встановив, що коефіцієнт поглинання пропорційний концентрації поглинаючої речовини, тобто:
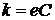 , ,
| (1.4) |
де 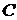 − концентрація речовини;
− концентрація речовини;
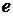 − коефіцієнт, не залежний від концентрації.
− коефіцієнт, не залежний від концентрації.
Закон Бера аналогічний закону Бугера-Ламберта. Закон Бугера-Ламберта розглядає зміну поглинання світлового потоку розчином постійної концентрації при зміні товщини поглинаючого шару, а закон Бера − зміну поглинання світлового потоку шаром постійної товщини при зміні концентрації.
Об'єднуючи формули (1.3) і (1.4), отримаємо рівняння основного закону фотометрії − закону Бугера-Ламберта-Бера:
 . .
| (1.5) |
Якщо концентрація виражена в молях на літр, а товщина шару − в сантиметрах, то коефіцієнт 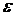 називають молярним коефіцієнтом поглинання; він є постійною величиною, залежною від довжини хвилі падаючого світла, природи розчиненої речовини, температури розчину, і відповідає світлопоглинанню молярного розчину аналізованої речовини.
називають молярним коефіцієнтом поглинання; він є постійною величиною, залежною від довжини хвилі падаючого світла, природи розчиненої речовини, температури розчину, і відповідає світлопоглинанню молярного розчину аналізованої речовини.
Чим більше значення , тим вища чутливість фотометричного метода.
Поглинання розчинами сильно залежить від довжини хвилі світла, що поглинається. Крива залежності коефіцієнта поглинання від довжини хвилі називається кривою спектрофотометрії. Ця крива охоплює не тільки область видимої частини спектру, яка використовується у візуальному фотометричному аналізі, але і ультрафіолетову та інфрачервону частини спектру.
Крива, що виражає графічно основний закон фотометрії має такий же вигляд, як і крива, приведена на рисунку 1.2. Відмінність полягає лише в тому, що оскільки в цьому випадку мова йде про розчини, по осі абсцис повинні бути нанесені концентрації, досліджувані при постійній товщині шару. Нахил кривої визначається поглинаючими властивостями речовини, тобто його коефіцієнтом поглинання.
Шляхом перетворення рівняння (1.5) можна вивести значення деяких фотометричних величин, з якими зазвичай доводиться мати справу.
Відношення інтенсивності світлового потоку, що пройшов через розчин, до інтенсивності падаючого світлового потоку у відсотках називають пропусканням і позначають буквою Т:
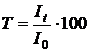 . .
| (1.6) |
Величина Т, що відноситься до товщини шару в 1 см, називається коефіцієнтом пропускання.
Логарифм відношення 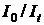 називається оптичною густиною (D):
називається оптичною густиною (D):
 . .
| (1.7) |
З цього рівняння виходить, що оптична густина D прямопропорційна концентрації речовини в розчині. Залежність між оптичною густиною і концентрацією може бути представлена графічно. Якщо дотримується закон Бугера-Ламберта-Бера, то отримуємо пряму лінію, що проходить через початок координат. Нахил її залежить від товщини шару і молярного коефіцієнта поглинання (рисунок 1.3, криві 1 та 2). Якщо за тих або інших обставин спостерігається відхилення від основного закону фотометрії, то залежність виражається кривою. Наприклад, на рисунку 1.3, крива 3 до концентрації 3 мкг/см3 спостерігається пряма пропорційність оптичної густини від концентрації, а при вищих концентраціях графік криволінійний.
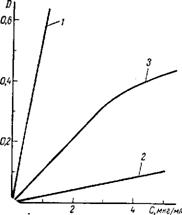
1 − дифенілкарбазидний комплекс хрому; 2 − роданідний комплекс молібдену; 3 − комплексу срібла з n-діетиламінобензиліденроданидом.
Рисунок 1.3 – Залежність оптичної густини розчинів від їх концентрацій
Об'єктивні помилки фотометрії. Джерелами помилок при фотометруванні можуть бути відхилення від закону Бугера-Ламберта-Бера і особливості виникаючого забарвлення.
1. Реакцію переведення визначуваного іона в забарвлене з'єднання можна представити рівнянням:
 , ,
| (1.8) |
де X − іон, який визначається в більшость випадків безбарвний або слабко-забарвлений;
R − реагент, забарвлений інакше, ніж сполука XR;
ХR – забарвлена сполука.
Розглянута реакція оборотна і константа дисоціації сполуки XR виражається рівнянням:
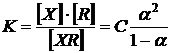 , ,
| (1.9) |
де С − концентрація забарвленої сполуки XR;
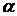 − ступінь дисоціації сполуки XR.
− ступінь дисоціації сполуки XR.
Інтенсивність забарвлення розчину, залежить від співвідношення концентрацій забарвлених і безбарвних частинок, змінюється із зміною загальної концентрації розчину, оскільки при цьому відбувається одночасна зміна ступеню дисоціації. В результаті при розбавленні або при концентрації розчинів спостерігаються відхилення від основного закону фотометрії.
2. Більшість забарвлених сполук чутливі до концентрації іонів водню. При зміні рН розчину відбувається зміна молярного коефіцієнта поглинання, а дуже часто зміна і кривої світлопоглинання. Як приклад на рисунку 1.4 приведені криві світлопоглинання галію з морином при рН 3,6 (крива 1) і рН 4,6 (крива 2). В цьому випадку змінюється тільки оптична густина. Для комплексу галію з метилтимоловим синім крива 3 знята при рН 1,8, а крива 4 при рН 6; тут при зміні рН спостерігається зміна і положення максимуму світлопоглинання, і оптичної густини.
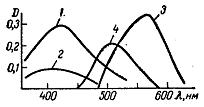
Рисунок 1.4 – Зміна кривих світлопоглинання комплексів галію з морином (1 і 2) і метилтимоловим (3, 4) залежно від рН
Залежність стійкості комплексу від рН зручно зображувати графічно. В більшості випадків із збільшенням рН оптична густина D спочатку збільшується (рисунок 1.5) – це пов’язано із зміцненням комплексу, що утворився. У визначеному інтервалі рН оптична густина стає постійною і саме цей інтервал є найбільш цікавим для аналітичного використання, так як тут не потребується суворого дотримання рН. При подальшому збільшенні рН оптична густина розчину знижується в результаті розкладу забарвленого комплексу і утворення гідроксиду.
1 – алюмінон-алюмінівий комплекс; 2 – кремніймоліденова гетерополікислота.
Рисунок 1.5 – Залежність оптичної густини від рН
3. Дуже часто відхилення від основного закону фотометрії викликається процесом комплексоутворення, наприклад:
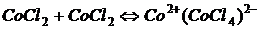 . .
| (1.10) |
Розчин СoCl2, синій при високих концентраціях, стає рожевим при розбавленні внаслідок утворення комплексу Со(СоСl4). Одночасно з цим відбувається зміна. Подібні зміни можуть відбуватись при комплексоутворенні за участі молекул розчинника, наприклад:
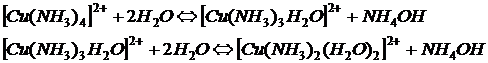
Всі ці іони мають різні молярні коефіцієнти поглинання. Тому з розбавленням розчину, який містить яскраво-сині 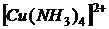 -іони, і у міру утворення блакитних іонів
-іони, і у міру утворення блакитних іонів  відбуватиметься зміна оптичної густини, пов'язана не тільки зі зміною концентрації, але і зі зміною молярних коефіцієнтів поглинання забарвлених іонів. Зміна оптичній густини може відбуватися також внаслідок введення в розчин сторонніх іонів. Наприклад, розчин Fe(NCS)3 при додаванні NаF знебарвлюється, оскільки відбувається утворення безбарвного комплексу [FeF6]3-.
відбуватиметься зміна оптичної густини, пов'язана не тільки зі зміною концентрації, але і зі зміною молярних коефіцієнтів поглинання забарвлених іонів. Зміна оптичній густини може відбуватися також внаслідок введення в розчин сторонніх іонів. Наприклад, розчин Fe(NCS)3 при додаванні NаF знебарвлюється, оскільки відбувається утворення безбарвного комплексу [FeF6]3-.
4. У більшості забарвлених сполук інтенсивність забарвлення змінюється з часом. У загальному випадку спочатку відбувається поступове зростання оптичної густини, оскільки реакції утворення забарвлених сполук йдуть в часі. Потім на певний інтервал часу забарвлення стабілізується і саме цей період використовується у фотометричному аналізі. Потім наступає явище «вицвітання» забарвлення − зменшення оптичної густини, пов'язане з дією кисню повітря, сонячного світла та інших чинників. Загальний характер оптичної густини розчину забарвленої сполуки в часі ілюструється на рисунку 1.6. В деяких випадках, наприклад, для купферонату ванадію, період дозрівання забарвлення дуже малий і воно стабільне протягом 30−40 хвилин. Забарвлення фосфоромолібденового комплексу, навпаки, поволі дозріває, протягом 40−60 хвилин, а потім дуже довгий час залишається стабільною. Тому завжди при фотометричних дослідженнях необхідно попередніми дослідами встановити інтервал часу стабільності досліджуваної забарвленої сполуки.
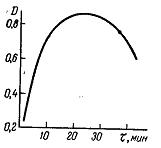
Рисунок 1.6 – Зміна оптичної густини розчину забарвленої сполуки в часі
5. Інтенсивність забарвлення, а отже, і поглинання залежать від температури.
Поглинання деяких сполук, наприклад, роданідного комплексу вольфраму, зростає з підвищенням температури, а для роданідного комплексу молібдену сполуки йоду з крохмалем спостерігається зворотна залежність.
Отже, при фотометруванні необхідно підтримувати постійну температуру; проте вплив температури в більшості випадків незначний, і тому вона повинна підтримуватися постійною в межах ±(1 – 3)°С. Відмінність залежності поглинання від температури використовується в термоспектрофотометричному методі аналізу.
6. Відхилення від закону Бугера-Ламберта-Бера можуть бути викликані і сторонніми речовинами, присутніми в розчині. Вплив сторонніх речовин можна розділити на декілька типових випадків.
Стороння речовина забарвлена. В цьому випадку на забарвлення досліджуваної речовини накладається забарвлення сторонньої речовини. Якщо це забарвлення постійне, то калібрувальний графік не проходить через початок координат, але може бути використаний для визначення досліджуваної речовини. Якщо забарвлення сторонньої речовини непостійне, то в цьому випадку іноді можна вибрати відповідну довжину хвилі, яку стороння речовина не поглинає, а досліджувана поглинає. Іноді такі розчини можна фотометрувати методами диференціальної фотометрії. Найбільш радикальним методом придушення шкідливого впливу сторонньої речовини є видалення його з розчину тим або іншим шляхом (екстрагуванням, окисленням, осадженням та ін.).
Стороння речовина реагує з реагентом. В цьому випадку утворюється змішане забарвлення досліджуваної і сторонньої речовини. Якщо концентрація сторонньої речовини постійна і забарвлення не дуже інтенсивне, то можна вести фотометричний аналіз, але калібрувальний графік починатиметься не від нуля. Як і в першому випадку, можна використовувати фотометрування при відповідній довжині хвилі. В деяких випадках можна вести фотометричний аналіз, вимірюючи спочатку сумарну оптичну густину, а потім тим або іншим способом, зруйнувавши одну із сполук, визначити оптичну густину сполуки, що залишилася, потім за різницею можна знайти концентрацію зруйнованої сполуки. Як і в першому випадку, найбільш радикальним способом є видалення сторонньої речовини.
Стороння речовина впливає на забарвлення. Це найбільш складний випадок, оскільки дуже часто невідома природа такого впливу. Іноді цей вплив пов'язаний із зміною іонної сили розчину або з утворенням змішаних комплексів невідомого і складного складу. Найбільш радикальним виходом в даному випадку є видалення речовини, що заважає.
7. Утворення забарвленої сполуки і інтенсивність її забарвлення дуже часто залежать від таких умов, як кількість реактиву, що додається, порядок додавання, концентрація реагенту та інші.
На рисунку 1.7 приведена залежність оптичної густини розчину роданідного комплексу молібдену від кількості доданого роданіду калія. Із збільшенням кількості роданіду калія оптична густина зростає до деякої межі, після якої надлишок реагенту більше не впливає на забарвлення. Інші реагенти можуть поводитися інакше, наприклад, додавання надлишку їх може приводити до зменшення оптичної густини. Отже, кількість реактивів, що додаються, необхідно строго контролювати, інакше можливе відхилення від основного закону фотометрії.
Рисунок 1.7 – Залежність оптичної густини розчину роданідного комплексу молібдену від кількості доданого роданіду калія
Дата добавления: 2018-05-13; просмотров: 549; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!