Исследование уравнения Ван-дер-Ваальса. Критическое состояние в-ва
1.  Графикуравнения Ван-дер-Ваальса в системе координат p,V представляет собой кубическую параболу. На рис.42 показана кривая для 1 моля воды при 373 К. Любому значению давления
Графикуравнения Ван-дер-Ваальса в системе координат p,V представляет собой кубическую параболу. На рис.42 показана кривая для 1 моля воды при 373 К. Любому значению давления 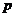 может соответствовать одно (точки 1 и 3) или три значения объема (точки 2). В двух случаях, когда давление соответствует точке максимума c или точке минимума e кривой, из трех значений объема
может соответствовать одно (точки 1 и 3) или три значения объема (точки 2). В двух случаях, когда давление соответствует точке максимума c или точке минимума e кривой, из трех значений объема  два одинаковы.
два одинаковы.
Уравнение Ван-дер-Ваальса есть уравнение кубическое относительно . Поэтому значения объемов в общем случае являются корнями этого уравнения при определенном наборе параметров v, p, T, R, a, b.
2. Сравнение уравнения Ван-дер-Ваальса с уравнением Клапейрона-Менделеева. Преобразуем уравнение к виду, удобному для сравнения: 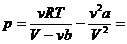
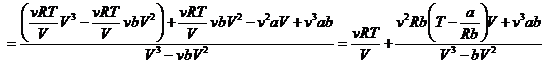 (14.1)
(14.1)
Комбинацию констант a| Rb = TБ называют температурой Бойля.При температуре газа, равной температуре Бойля, Т = ТБ, числитель дроби принимает минимальное абсолютное значение. Газ при этой температуре ближе всего подходит по своим свойствам к идеальному.
В зависимости от знака разности Т – ТБ давление реального газа с уменьшением объема изменяется быстрее (Т > ТБ) или медленнее (Т < ТБ), чем идеального газа. При Т > ТБ скобка положительна, давление реального газа растет быстрее, силы отталкивания преобладают над силами притяжения. При Т < ТБ скобка отрицательна, и если числитель дроби отрицателен, то с уменьшением объема давление реального газа растет медленнее, чем идеального. Силы притяжения преобладают над силами отталкивания.
3. Пересыщенный пар и перегретая жидкость. При совмещении экспериментальной изотермы с теоретической наблюдаются расхождения (рис.43). Проанализируем их, двигаясь по изотерме в направлении сжатия газа (справа налево по рис.43). Участки ab и fg опытной кривой совпадают с изотермой Ван-дер-Ваальса. Участок ab соответствует газообразному состоянию сжимаемого вещества, а участок fg – жидкому.
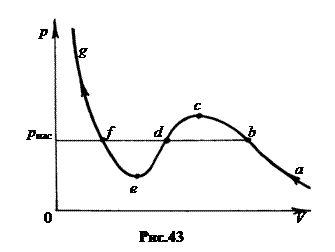 Состояния, соответствующие участку cde теоретической изотермы в природе существовать по-видимому не могут, так как с уменьшением давления объем вещества не увеличивается, а уменьшается, и наоборот. Поэтому вещество должно находиться здесь в нестабильном состоянии. Оно должно самопроизвольно или коллапсировать в точку e, или раздуваться в точку c.
Состояния, соответствующие участку cde теоретической изотермы в природе существовать по-видимому не могут, так как с уменьшением давления объем вещества не увеличивается, а уменьшается, и наоборот. Поэтому вещество должно находиться здесь в нестабильном состоянии. Оно должно самопроизвольно или коллапсировать в точку e, или раздуваться в точку c.
Участок bc, продолжающий ветвь газообразного состояния ab, соответствует также газообразному состоянию, хотя и несколько ненормальному. Давление газа в любом из этих состояний участка bc больше давления насыщенного пара рнас.
 Такие состояния могут быть реализованы, например, путем быстрого охлаждения тщательно очищенных паров до температуры ниже температуры конденсации. Если нет центров конденсации, пар оказывается пересыщенным. Количество его больше, чем нужно для того, чтобы он был насыщенным.
Такие состояния могут быть реализованы, например, путем быстрого охлаждения тщательно очищенных паров до температуры ниже температуры конденсации. Если нет центров конденсации, пар оказывается пересыщенным. Количество его больше, чем нужно для того, чтобы он был насыщенным.
Состояния, соответствующие участку ef, называются перегретой жидкостью. Их реализовать еще труднее, чем пересыщенный пар. Состояния bc и ef называют метастабильными.
4. Правило Максвелла. Анализ уравнения Ван-дер-Ваальса не позволяет ответить на вопрос: где должна проходить опытная горизонтальная изобара bdf двухфазного состояния системы?
В 1875 г. Джеймс Максвелл указал способ определения положения этой прямой: - так как переход из состояния «b» в состояние «f», независимо от того, будет ли он совершаться по теоретической кривой изотермы, или по опытной изобаре, должен сопровождаться одной и той же работой сжатия вещества, то изобара должна проходить так, чтобы площади криволинейных фигур S1 и S2 (рис.44) в системе p,V, были одинаковыми.
5. Критическое состояние вещества. С повышением температуры длина опытной изобары уменьшается и при Т = Тk вырождается в точку k (Т. Эндрюс, рис.45).
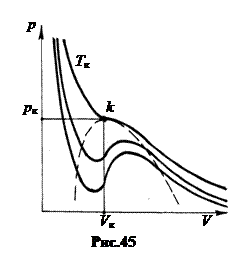 Кубическая парабола уравнения Ван-дер-Ваальса в этой точке испытывает перегиб, то есть имеет экстремум, для которого первая и вторая производные от p по V равны нулю. Используя эти условия, можно найти параметры pk, Vk и Тk, соответствующие критическому состоянию вещества.
Кубическая парабола уравнения Ван-дер-Ваальса в этой точке испытывает перегиб, то есть имеет экстремум, для которого первая и вторая производные от p по V равны нулю. Используя эти условия, можно найти параметры pk, Vk и Тk, соответствующие критическому состоянию вещества.
Уравнение Ван-дер-Ваальса: 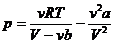 . (14.2)
. (14.2)
Первая производная от p по V:  . (14.3)
. (14.3)
Вторая производная от p по V: 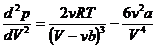 . (14.4)
. (14.4)
В критической точке получаем систему из трех уравнений с тремя неизвестными pk, Vk, Тk.
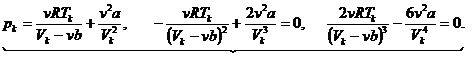 (14.5)
(14.5)
 Разнеся члены второго и третьего уравнений по разные стороны знака равенства и разделив второе на третье, получаем Vk = 3vb. Из второго уравнения, подставив Vk, получаем:
Разнеся члены второго и третьего уравнений по разные стороны знака равенства и разделив второе на третье, получаем Vk = 3vb. Из второго уравнения, подставив Vk, получаем: 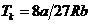 . И из первого:
. И из первого:  . Эти выражения позволяют находить из опытных значений pk, Vk, Тk поправки a и b: b=Vk| 3v,
. Эти выражения позволяют находить из опытных значений pk, Vk, Тk поправки a и b: b=Vk| 3v, 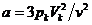 . (14.6)
. (14.6)
В таблице 9 приведены опытные значения критических параметров pk, Vk, Тk некоторых веществ и вычисленные по ним a и b.
В критическом состоянии изобара конденсации вырождается в точку. Это значит, что исчезает переход из газообразного состояния в жидкое и наоборот. При Т > Тk кинетическая энергия движения молекул больше их энергии связи, поэтому силы молекулярного сцепления не могут удержать вещество в конденсированном состоянии.
Критическое состояние – это не газ и не жидкость. Сжимаемость вещества в критическом состоянии бесконечно велика.  . (14.7)
. (14.7)
 При одном и том же давлении pk разные области вещества могут сильно отличатся концентрациями частиц. Поэтому вещество в критическом состоянии очень неоднородно. Оптически это проявляется в сильном помутнении обычно прозрачных веществ – критической опалесценции.
При одном и том же давлении pk разные области вещества могут сильно отличатся концентрациями частиц. Поэтому вещество в критическом состоянии очень неоднородно. Оптически это проявляется в сильном помутнении обычно прозрачных веществ – критической опалесценции.
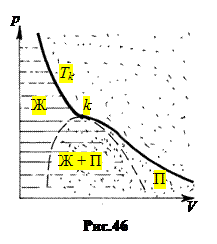
6. Пар и газ. Эти термины имеют разный смысл. Пар можно превратить в жидкость изотермическим сжатием, а газ – нет (рис.46). Пар – это газообразное состояние вещества при температуре ниже критической, Т < Тk. Газообразная вода в атмосфере – это пар (Тk у воды 374,15°С). А кислород и азот в атмосфере – газы, так как их критические температуры (- 129°С у кислорода и – 147°С у азота) много выше температуры атмосферы. Углекислота СО2 при Т > 31,1°С – газ, а при Т < 31,1°С – пар.
7. Параметры уравнения Ван-дер-Ваальса a и b, строго говоря, не являются константами. Они заметно изменяются с температурой. Так у аргона, например, при Т = 424 К b = 61×10- 6 м3|моль, а = 0,19 Дж×м3|моль2. С ростом температуры величины а и b уменьшаются. При Т = 546 К b = 41,0×10- 6 м3|моль, а = 0,14 Дж×м3|моль2.
Кроме того, опыт показывает, что лучше выполняется условие Vk = 2vb, а не Vk = 3vb. Это говорит о том, что уравнение Ван-дер-Ваальса приближенное, его использование для количественных расчетов требует осторожности.
8. Другие уравнения состояния реальных газов. Из-за недостаточной точности уравнения Ван-дер-Ваальса вскоре стали появляться другие варианты уравнения состояния реальных газов. Отметим два из них.
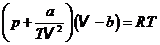 . Марселен Бертло.
. Марселен Бертло. 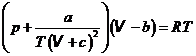 . Рудольф Клаузиус
. Рудольф Клаузиус
Уравнение Бертло при умеренных давлениях лучше согласуется с опытом, но вблизи критической точки не имеет преимуществ перед уравнением Ван-дер-Ваальса.
 Уравнение Клаузиуса лучше соответствует опыту в более широком диапазоне давлений и температур. Но это достигается ценой введения третьей эмпирической константы с, определяемой эмпирически.
Уравнение Клаузиуса лучше соответствует опыту в более широком диапазоне давлений и температур. Но это достигается ценой введения третьей эмпирической константы с, определяемой эмпирически.
9. Измерение критических параметров. В первую очередь определяется критическая температура Тk. Назовем 3 метода измерений Тk.
а. Метод Эндрюса, 1869 г. Вещество изотермически сжимается при последовательно повышающихся от опыта к опыту температурах. Постепенно «нащупывается» то значение Тk, при котором изобара вырождается в точку. Это очень трудоемкий метод.
б. Метод исчезновения мениска, Каньярдела Тур, 1822. Вещество помещается в прозрачную ампулу при температуре ниже критической и нагревается. Если количество вещества мало отличается по критическому объему от объема ампулы, то жидкость при нагревании не испарится, и не заполнит всю ампулу. При достижении Т = Тk мениск исчезает.
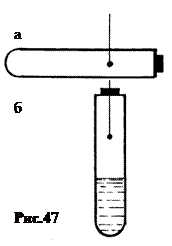
в. Метод А. Надеждина,1885. Пустая металлическая ампула уравновешивалась так, что на горизонтальной оси принимала горизонтальное положение (рис.47-а). После заливки порции вещества ампула поворачивалась вертикально (рис.47-б). В таком положении она нагревалась. Когда температура достигала критической, вещество равномерно распределялось по всему объему, и ампула поворачивалась в горизонтальное положение.
Критическое давление обычно находится изотермическим сжатием вещества при критической температуре по точке перегиба. Молярный критический объем находят чаще всего подбором количества вещества в ампуле.
Эффект Джоуля-Томсона.
1. Внутренняя энергия реального газа. Вследствие хаотичного движения молекулы реального газа обладают такой же кинетической энергией, как и молекулы идеального газа. Величина этой кинетической энергии 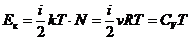 . Но в отличие от идеального газа молекулы реального газа еще взаимно притягиваются, чем обусловлена их потенциальная энергия Ек. Изменение потенциальной энергии газа в случае его изотермического расширения равно работе, которую должны совершить внешние тела по преодолению сил внутреннего давления.
. Но в отличие от идеального газа молекулы реального газа еще взаимно притягиваются, чем обусловлена их потенциальная энергия Ек. Изменение потенциальной энергии газа в случае его изотермического расширения равно работе, которую должны совершить внешние тела по преодолению сил внутреннего давления. 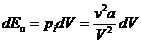 . (15.1)
. (15.1)
Потенциальная энергии реального газа 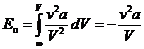 . (15.2)
. (15.2)
 Итак, внутренняя энергия реального газа равна
Итак, внутренняя энергия реального газа равна 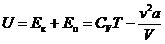 . (15.3)
. (15.3)
2. Опыт Джоуля-Томсона состоит в пропускании газа через пористую перегородку в теплоизолированной трубе с большим перепадом давления. Он был реализован в совместных экспериментах Дж.Джоуля и У.Томсона в 1852 – 62 г.
В теплоизолированной трубе слева направо (по рис.48) двигаются поршни S1 и S2. Газ из левой части трубы под давлением р1 продавливается (дросселирует) через пористую перегородку А в правую часть, в которой давление р2 << р1. Давления р1 и р2 постоянны. Чтобы сохранять перепад давления р1 - р2 большим, стенка S2 должна двигаться быстрее стенки S1.
Оказалось, что в одних случаях температура газа понижалась, в других – повышалась. Это изменение температуры называют эффектом Джоуля – Томсона.
3. Теория опыта Джоуля – Томсона. Определим величину и знак изменения температуры газа DT = T2 - T1. Пусть 1 моль газа занимал перед дросселированием объем V1 в левой части трубы под давлением p1, а после дросселирования – объем V2 под давлением p2в правой части. Причем р1 >> р2, а V2 >> V1. Работа по перемещению левого поршня S1 совершается внешними телами и равна A1 =p1V1. Работа по перемещению правого поршня S2 совершается газом над внешними телами и равна A2 =p2V2.
Работа, совершаемая внешними телами над газом, в целом есть A1 - A2 = p1V1 - p2V2. Эта работа идет на увеличение внутренней энергии газа.
A1 - A2 = U2 - U1 или A1 + U1 = A2 + U2, или U1 + p1V1 = U2 + p2V2 . (15.4)
Подставим сюда выражение (15.3) для внутренней энергии реального газа и давление p из уравнения Ван-дер-Ваальса по формуле (14.2).
 . (15.5)
. (15.5)
Равенство упрощается, если учесть, что в правой части объем V2 велик, и поэтому членами 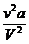 и vb можно пренебречь:
и vb можно пренебречь: 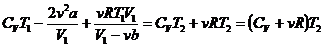 . (15.6)
. (15.6)
Преобразуем 3-й член:  и подставляем. (15.7)
и подставляем. (15.7)
 , или
, или  . Отсюда
. Отсюда  . (15.8)
. (15.8)
 4. Температура инверсии. Знак изменения температуры зависит от соотношения членов в скобках. Могут быть ситуации, когда DT = 0. В этом случае
4. Температура инверсии. Знак изменения температуры зависит от соотношения членов в скобках. Могут быть ситуации, когда DT = 0. В этом случае 
 . (15.9)
. (15.9)
Температура Т1, при которой эффект Джоуля – Томсона не наблюдается (DT = 0), зависит от величины начального мольного объема газа V1, который формально не может быть меньше поправки b. С ростом начального объема V 1 температура Т1 нулевого эффекта стремится к пределу 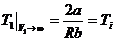 , (15.10)
, (15.10)
 который называется температурой инверсии.
который называется температурой инверсии.
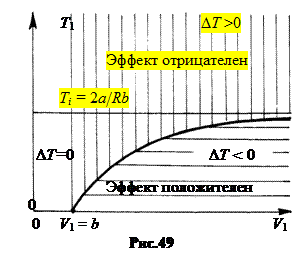
Если температура газа при дросселировании понижается, DT < 0, говорят эффект положителен. Если газ при дросселировании нагревается, DT > 0, говорят, эффект отрицателен.
 Точки кривой на графике (рис.49) соответствуют температуре Т1, при переходе через которую при данном начальном объеме V1 знак эффекта Джоуля – Томсона меняется. Если Т1 > Тi = 2a| Rb, то знак эффекта уже не зависит от начального объема, DT > 0, эффект только отрицателен. Величина температурного перепада DT изменяется с величиной начальных параметров Т1 и V 1 (рис.50).
Точки кривой на графике (рис.49) соответствуют температуре Т1, при переходе через которую при данном начальном объеме V1 знак эффекта Джоуля – Томсона меняется. Если Т1 > Тi = 2a| Rb, то знак эффекта уже не зависит от начального объема, DT > 0, эффект только отрицателен. Величина температурного перепада DT изменяется с величиной начальных параметров Т1 и V 1 (рис.50).
Чтобы найти оптимальный объем V 1опт, при котором положительный эффект максимален, надо исследовать DT на экстремум по V1: 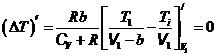 . (15.11)
. (15.11)
Отсюда,
 , или
, или  . (15.12)
. (15.12)
Здесь k = T1| Ti . Так как DT < 0, то k < 1. Из уравнения (15.1) находится оптимальный начальный объем.
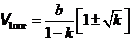 . (15.13)
. (15.13)
Так как минус в скобках приводит к V 1 < b, то остается один корень.
 . (15.14)
. (15.14)
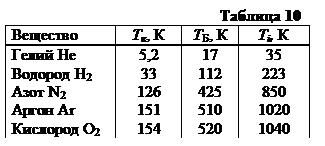 5. Характеристические температуры реальных газов. Это температуры критическая Тк, Бойля ТБ и инверсии Тi (таблица 10).
5. Характеристические температуры реальных газов. Это температуры критическая Тк, Бойля ТБ и инверсии Тi (таблица 10).

Все температуры однозначно определяются поправками Ван-дер-Ваальса a и b.
Знание этих температур позволяет прогнозировать степень отклонения реальных газов от идеальных при разных температурах и выбирать оптимальные условия для сжижения газов.
Изменение температуры газа при его адиабатном дросселировании объясняется тем, что при расширении газа увеличивается расстояние между молекулами, а, следовательно, совершается работа против сил взаимодействия между ними. Эта работа совершается за счет кинетической энергии молекул (расширение адиабатное), что и приводит к изменению температуры газа.
6. Сжижение газов. Технология сжижения сводится к охлаждению газа до температур ниже критической и сжатию газа при этих температурах.
а. Метод Линде, 1895. Поскольку за один цикл адиабатного дросселирования не удается понизить температуру газа до критической, немецкий инженер Линде предложил дросселировать газ многократно, понижая постепенно его температуру в теплообменнике на (рис.51).
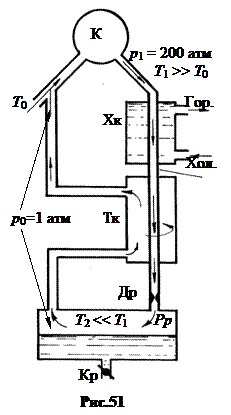 Компрессор К всасывает газ при комнатной температуре Т0 и давлении р0 = 1 атм и сжимает его до давления р1 = 200 атм. Газ при этом нагревается. Пройдя водяной холодильник Хк, он вновь остывает до температуры Т0. Пройдя дроссель Др, после которого давление падает с 200 до 1 атм, газ охлаждается до Т2 < T1 и попадает в резервуар Рр. Но после первого прохода газ еще не охладился до температуры ниже критической. Поэтому он возвращается в компрессор К через теплообменник Тк , где охлаждает вторую порцию газа до Т < T0. После многократного прохождения газа через теплообменник его температура понижается постепенно ниже критической, после чего при дросселировании газ частично конденсируется и накапливается в резервуаре Рp. Через кран Кр конденсат периодически сливается в сосуды Дьюара.
Компрессор К всасывает газ при комнатной температуре Т0 и давлении р0 = 1 атм и сжимает его до давления р1 = 200 атм. Газ при этом нагревается. Пройдя водяной холодильник Хк, он вновь остывает до температуры Т0. Пройдя дроссель Др, после которого давление падает с 200 до 1 атм, газ охлаждается до Т2 < T1 и попадает в резервуар Рр. Но после первого прохода газ еще не охладился до температуры ниже критической. Поэтому он возвращается в компрессор К через теплообменник Тк , где охлаждает вторую порцию газа до Т < T0. После многократного прохождения газа через теплообменник его температура понижается постепенно ниже критической, после чего при дросселировании газ частично конденсируется и накапливается в резервуаре Рp. Через кран Кр конденсат периодически сливается в сосуды Дьюара.
Метод Линде прост, в машине нет движущихся частей (за исключением компрессора) и поэтому нет проблемы смазки. Но он малоэффективен.
б. Метод Клода, 1902 г. Французский инженер Клод заменил дроссель цилиндром с поршнем. Благодаря этому расширение газа сопровождается совершением им работы по перемещению поршня. Температура газа падает быстрее. Поэтому машина Клода экономичнее.
ГЛАВА 4. ЖИДКОСТИ
Объемные свойства жидкостей
1. Основное отличие жидкостей от газов состоит в том, что жидкости сохраняют свой объем и имеют свободную поверхность. Поэтому свойства жидкостей разделяют на объемные и поверхностные. В этом параграфе рассматриваются объемные свойства.
 2. Сжимаемость. Изотермический коэффициент сжимаемости жидкостей c = -(dV | dp)T | V по сравнению с идеальными газами, для которых c = 1| p, очень мал (Таблица11).
2. Сжимаемость. Изотермический коэффициент сжимаемости жидкостей c = -(dV | dp)T | V по сравнению с идеальными газами, для которых c = 1| p, очень мал (Таблица11).
При нормальном атмосферном давлении p = 105 Па сжимаемость идеальных газов c = 10-5 Па-1. Сжимаемость жидкостей на 5 порядков меньше.
Например, абсолютное уменьшение объема 1 м3 воды по давлением 1 атм составляет DV = cV×Dp = 5,1×10-10×1×105 = 5,1×10-5 м3 =51 см3. Столь незначительное сжатие приводит, тем не менее, к понижению уровня мирового океана на 30 м.
Если перейти в формуле для c к конечным разностям, что допустимо при не очень больших давлениях, при которых c остается постоянной, то можно получить формулу, удобную для практических расчетов.  . Если V0 – объем жидкости при некотором давлении p0, то DV = V - V0, где V – объем жидкости при давлении p. Отсюда
. Если V0 – объем жидкости при некотором давлении p0, то DV = V - V0, где V – объем жидкости при давлении p. Отсюда
V - V0 = - c V0 (p - p0); Þ V = V0 [1 -c(p - p0)] . (16.1)
С увеличением давления p коэффициент c уменьшается, и при очень больших давлениях p > 108 Па сжимаемости всех жидкостей становятся практически одинаковыми.
Поскольку с повышением температуры жидкость становится более рыхлой, то нагревание жидкостей приводит к увеличению их коэффициента сжимаемости c.
| Таблица 12 | ||
| Жидкость | Интервал темп-р, °C | b, K-1 |
| Ацетон Вода Глицерин Ртуть Спирт эт. Эфир эт. | -95÷56,5 0÷100 -17÷290 -39÷357 -114÷78 -116÷34 | 1,487×10-3 0,208×10-3 0,505×10-3 0,181×10-3 1,100×10-3 1,650×10-3 |
3. Тепловое расширение. По сравнению с идеальными газами, для которых изобарический коэффициент теплового расширения b = (dV | dT)p| V равен 1/T и при T = 273 К составляет b = 1| 273 = 3,7×10-3 K-1, тепловое расширение жидкостей отличается уже меньше (Таблица 12).
У воды имеется температурная аномалия, состоящая в том, что плотность воды максимальна не в точке замерзания t = 0°C, а при t = 4°C. Дело в том, что молекулы H2O частично ассоциируют в группы (H2O)2, (H2O)3, удельный объем которых различен. При разных температурах соотношение концентраций этих групп разное. При 4°С соотношение ассоциатов таково, что плотность упаковки молекул H2O максимальна. (При t = 0°C r = 999,841 кг/м3, а при t = 4°C r = 999,973 кг/м3).
В общем случае коэффициент b меняется с температурой. Но на линейных участках, где коэффициент b стабилен, в формуле для b можно перейти к конечным разностям.
 . (16.2)
. (16.2)
Если в качестве отсчетного брать объем V0 при T0 = 273 K, то DT = t°C, и
DV = V - V0 = b V0 t Þ V = V0 =(1 + bt). (16.3)
4. Теплоемкость жидкостей зависит от температуры, причем у разных жидкостей по-разному. У большинства жидкостей с ростом T теплоемкость увеличивается, у некоторых, например, у ртути – уменьшается. Эти колебания теплоемкости могут доходить до 20%.
У воды теплоемкость минимальна при 40°C. При изменении температуры в обе стороны она растет. При t = 0°C c = 4,212×103 Дж/кг×К, при t = 40°C c = 4,174×103 Дж/кг×К, а при t = 100°C c = 4,220×103 Дж/кг×К. Но в целом в диапазоне температур от 0°C до 100°C эти колебания в пределах 1%.
Как и у газов, у жидкостей различают две теплоемкости Cp и CV. Разность Cp - CV = vR также численно равна работе изобарного расширения при нагревании на 1 К. Величина ее у разных жидкостей различна и может быть больше или меньше vR.
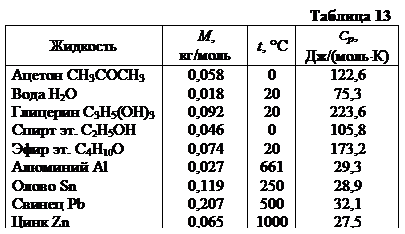 В таблице 13 приведены значения молярной теплоемкости Cp для некоторых жидкостей. Интересно, что у жидких металлов молярная теплоемкость Cp приближается к 3R@25 Дж|(моль×К).
В таблице 13 приведены значения молярной теплоемкости Cp для некоторых жидкостей. Интересно, что у жидких металлов молярная теплоемкость Cp приближается к 3R@25 Дж|(моль×К).
5. Строение жидкостей. Молекулы в жидкостях находятся значительно ближе друг к другу, чем в газах. Если в газах силы межмолекулярного взаимодействия играют заметную роль лишь при низких температурах и значительных давлениях, то в жидкостях эти силы являются преобладающими.
Величина pi = a| V 2 в жидкостях много больше внешнего давления p. Для воды, например, pi = 1,1×109 Па ( = 11000 атм). Поэтому в уравнении Ван дер Ваальса для жидкостей внешними силами можно пренебречь. 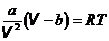 . (16.4)
. (16.4)
Внутреннее давление pi = a| V 2 можно определить путем измерения работы перехода молекул жидкости через поверхностный слой в газ. Такой выход молекул есть процесс испарения, а работа выхода найдется через величину теплоты испарения.
В расположении молекул жидкости наблюдается так называемый ближний порядок. Это значит, что по отношению к любой молекуле расположение ближайших к ней соседей является упорядоченным. Но по мере удаления от данной молекулы нарушения в регулярности накапливаются, и довольно быстро порядок в расположении молекул исчезает. Поэтому жидкости изотропны.
Теория строения жидкостей до сих пор разработана недостаточно и во многих случаях носит полуэмпирический характер. Согласно Якову Френкелю, разрабатывавшему теорию жидкого состояния в 30-х годах 20 в., каждая молекула колеблется около своего положения равновесия. Время от времени молекула меняет место равновесия, скачком перемещаясь в новое положение, отстоящее от прежнего на расстояние порядка диаметра молекул.
Среднее время пребывания молекул в одном месте различно у разных жидкостей и уменьшается с ростом температуры T. Чем выше T, тем чаще перескакивают молекулы. Поэтому с ростом T в жидкостях увеличивается коэффициент диффузии и уменьшается вязкость.
С увеличением T растет средняя энергия теплового движения молекул. Это проявляется в увеличении амплитуды колебания молекул около положения равновесия. Среднее расстояние между центрами молекул с ростом температуры T также увеличивается. Жидкость разрыхляется, ее объем растёт, плотность уменьшается.
6. Диффузия в жидкостях. В газах коэффициенты переноса определяются длиной свободного пробега молекул  . Но в жидкостях понятие длины свободного пробега молекул теряет смысл. Расстояние межмолекулярного скачка в жидкостях много меньше величины в газах. Поэтому и коэффициент диффузии в жидкостях много меньше, чем в газах.
. Но в жидкостях понятие длины свободного пробега молекул теряет смысл. Расстояние межмолекулярного скачка в жидкостях много меньше величины в газах. Поэтому и коэффициент диффузии в жидкостях много меньше, чем в газах.
| Таблица 14 | |||
| Диффундирующее вещество | Среда | T, K | D, м2/с |
| Газ H2 Сахар C12H22O11 Соль NaCl Золото Au тв. | Газ O2 Вода жидк. Вода жидк. Свинец Pb тв. | 273 293 283 293 | 0,7×10-4 0,3×10-9 1,1×10-9 4×10-14 |
В таблице 14 сравниваются коэффициенты диффузии, измеренные в трех средах – в газах, в жидкостях и твердых телах. Величина D в жидкостях на 5 порядков меньше, чем в газах, а в твердых телах на 5 порядков меньше, чем в жидкостях.
По Френкелю, коэффициент диффузии в жидкостях растет с температурой T по закону D = B×exp(- w| kT). (16.5)
 Здесь B – параметр, зависящий от свойств жидкости, w – энергия, необходимая для скачка молекулы (энергия активации), w @ (2 ¸ 3)×10-20 Дж, k = 1,38×10-23 Дж/K – постоянная Больцмана.
Здесь B – параметр, зависящий от свойств жидкости, w – энергия, необходимая для скачка молекулы (энергия активации), w @ (2 ¸ 3)×10-20 Дж, k = 1,38×10-23 Дж/K – постоянная Больцмана.
7. Теплопроводность. Плотность упаковки молекул жидкости примерно на 3 порядка больше плотности упаковки молекул газа. Но примерно во столько же раз расстояние скачка молекул жидкости меньше длины свободного пробега молекул газа. Поэтому коэффициенты теплопроводности газов и жидкостей-диэлектриков отличаются в пределах 1 порядка (Таблица 15).
Как видно из таблицы, теплопроводность жидкостей, обладающих электронной проводимостью (жидкие металлы) на 1-2 порядка больше теплопроводности жидкостей-диэлектриков.
С ростом температуры теплопроводность жидкостей уменьшается. У многих жидкостей эта зависимость c от T приближается к линейной. c = cпл [1 - a(t - tпл)]. (16.6)
Здесь cпл – теплопроводность жидкости в точке плавления tпл, a – коэффициент.
При опытном измерении теплопроводности жидкостей одной из важных задач является устранение конвекции. Обычно это достигается нагреванием жидкости сверху. Тогда легкие слои находятся вверху, более тяжелые – внизу. Подавлению конвекции способствует и уменьшение толщины слоя жидкости, через который проходит тепловой поток.
8. Вязкость жидкостей по сравнению с газами очень велика и быстро уменьшается с ростом температуры (Таблица 16).
| Таблица 16 | |||||
| Жидкость | t, °C | h, мкПа×с | Жидкость | t, °C | h, мкПа×с |
| Ацетон Вода Глицерин Спирт этиловый | -20 +20 0 +20 -20 +20 +200 -20 0 +20 | 513 322 1788 1004 134×106 1499×103 216 2780 1780 1190 | Эфир этиловый Алюминий Олово Ртуть Свинец | 0 +20 +661 900 232 1300 -20 0 200 400 900 | 296 243 1190 920 1810 740 1855 1685 1052 2170 1230 |
 По Френкелю, вязкость жидкостей с ростом температуры T уменьшается по закону:
По Френкелю, вязкость жидкостей с ростом температуры T уменьшается по закону:
h = A×exp(w| kT)., (16.7)
где A – параметр, зависящий от свойств жидкости.
Русский физик Алексей Бачинский предложил в 1912 г. формулу, в которой зависимость вязкости от температуры выражается в неявном виде через объем жидкости, 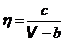 . (16.8)
. (16.8)
Здесь V – молярный объем жидкости, b – постоянная Ван-дер-Ваальса, c – параметр, зависящий от свойств жидкости.
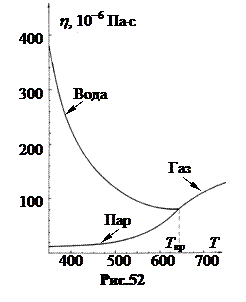
С ростом температуры вязкость жидкостей уменьшается, а вязкость равновесных паров растет. Если на одном графическом поле показать вязкость жидкости и ее равновесных паров, то в критической точке обе кривые сходятся (рис.52).
Сжатие жидкости приводит к росту вязкости. Например, увеличение давления от 1 до 20000 атмосфер приводит к увеличению вязкости воды на 6 порядков.
С понижением температуры вязкость жидкостей растет. Однако в 1938 г. Петр Капица открыл исключение из этого правила. Оно состоит в том, что вязкость жидкого гелия с приближением его температуры к абсолютному нулю не только не увеличивается, но полностью исчезает.
Это явление называют сверхтекучестью жидкого гелия. Оно имеет квантовую природу.
 §17. Поверхностные свойства жидкостей
§17. Поверхностные свойства жидкостей
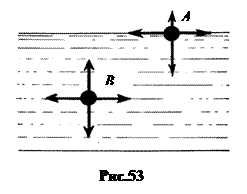
1. Поверхностный слой. Рассмотрим силы, действующие на молекулу жидкости в глубине ее и в плоском поверхностном слое (рис.53).
Силы притяжения, действующие на молекулу B со стороны окружающих молекул, в среднем одинаковы по всем направлениям.
Этого нельзя сказать о силах притяжения, действующих на молекулу А поверхностного слоя. Силы, действующие в плоскости поверхности, приблизительно одинаковы. Поэтому молекула А в плоскости поверхности находится в безразличном равновесии. Но силы притяжения со стороны молекул жидкости, лежащих ниже молекулы А , много больше сил притяжения со стороны молекул газа, лежащих выше молекулы А . Поэтому молекула А в целом притягивается к массе жидкости. И в таком положении находится каждая молекула поверхностного слоя.
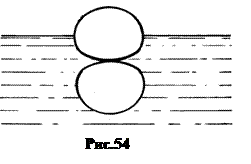 Благодаря разнице в силах притяжения со стороны молекул жидкости и со стороны молекул газа формируется поверхностный слой жидкости, отделяющий жидкое состояние вещества от газообразного. Поскольку каждая молекула поверхностного слоя притягивается нижерасположенными молекулами жидкости, то она смещается по направлению к молекулам жидкости до тех пор, пока силы притяжения не уравновесятся силами отталкивания (рис.54).
Благодаря разнице в силах притяжения со стороны молекул жидкости и со стороны молекул газа формируется поверхностный слой жидкости, отделяющий жидкое состояние вещества от газообразного. Поскольку каждая молекула поверхностного слоя притягивается нижерасположенными молекулами жидкости, то она смещается по направлению к молекулам жидкости до тех пор, пока силы притяжения не уравновесятся силами отталкивания (рис.54).
Поверхностный слой как бы подпрессовывает жидкость в целом и обладает избыточной по сравнению с остальной массой жидкости энергией. Эту энергию называют поверхностной. Силы притяжения между молекулами поверхностного слоя вызывают стремление жидкости сократить свою поверхность.
 Переход молекулы из глубины жидкости в поверхностный слой требует совершения работы против действующих в поверхностном слое сил. Эта работа совершается за счет кинетической энергии молекул. Поскольку молекулы жидкости обладают разной кинетической энергией, то среди молекул поверхностного слоя всегда найдутся такие, энергия которых достаточна для совершения работы по преодолению Вандерваальсовых сил притяжения. В результате молекула может перейти в газ. Поэтому поверхностный слой не мономолекулярен. Он разрыхлен тем больше, чем дальше находятся молекулы от основной массы жидкости (рис.55).
Переход молекулы из глубины жидкости в поверхностный слой требует совершения работы против действующих в поверхностном слое сил. Эта работа совершается за счет кинетической энергии молекул. Поскольку молекулы жидкости обладают разной кинетической энергией, то среди молекул поверхностного слоя всегда найдутся такие, энергия которых достаточна для совершения работы по преодолению Вандерваальсовых сил притяжения. В результате молекула может перейти в газ. Поэтому поверхностный слой не мономолекулярен. Он разрыхлен тем больше, чем дальше находятся молекулы от основной массы жидкости (рис.55).
2. Поверхностное натяжение. Вандерваальсовые силы притяжения, действующие между молекулами поверхностного слоя, обычно называют силами поверхностного натяжения. У разных жидкостей они разные. Их величина характеризуется коэффициентом поверхностного натяжения s. Уясним его смысл.
 Если на поверхности жидкости мысленно выделить произвольный контур, то молекулы, лежащие по одну сторону линии контура, притягиваются к молекулам, лежащим по другую сторону (рис.56). Поскольку жидкость изотропна, то сила поверхностного натяжения, приходящаяся на определенный отрезок длины контура, не зависит от его ориентации. Величина этой силы, приходящаяся на единицу длины контура, равна численно коэффициенту поверхностного натяжения s. Единица s - Н|м.
Если на поверхности жидкости мысленно выделить произвольный контур, то молекулы, лежащие по одну сторону линии контура, притягиваются к молекулам, лежащим по другую сторону (рис.56). Поскольку жидкость изотропна, то сила поверхностного натяжения, приходящаяся на определенный отрезок длины контура, не зависит от его ориентации. Величина этой силы, приходящаяся на единицу длины контура, равна численно коэффициенту поверхностного натяжения s. Единица s - Н|м.
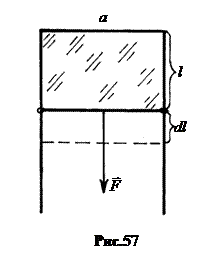 Найдем работу по увеличению площади поверхности пленки.
Найдем работу по увеличению площади поверхности пленки.
Пусть в рамке шириной a, на которой натянута жидкая пленка (рис.57), очень медленно перемещается перекладина под действием силы F = 2as на расстояние dl (2a потому, что у пленки две поверхности). Работа внешних сил перемещения перекладины идет на увеличение поверхностной энергии жидкости.
dA = dEп = Fdl = 2as×dl = sdS. (17.1)
Здесь dS = 2a×dl – приращение площади поверхности жидкости.
Отсюда Eп 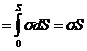 . (17.2)
. (17.2)
Итак, коэффициент s можно определить двумя способами.
а.  . Это сила поверхностного натяжения, отнесенная к единице длины контура.
. Это сила поверхностного натяжения, отнесенная к единице длины контура.
б.  . Это энергия поверхностного слоя, отнесенная к единичной площадке.
. Это энергия поверхностного слоя, отнесенная к единичной площадке.
| Опытные значения s для разных веществ Таблица 17 | |||||||
| Вещество | Пограничная среда | T, K | s, Н/м | Вещество | Пограничная среда | T, K | s, Н/м |
| Олово Ртуть Ртуть Висмут | Углекислый газ Пары ртути Воздух Углекислый газ | 526 288 291 1023 | 0,526 0,487 0,481 0,346 | Вода Масло каст. Скипидар Эфир этил. | Воздух Воздух Воздух Пары собств. | 288 291 291 293 | 0,073 0,033 0,027 0,016 |
3. Взаимодействие жидкости с другими телами. Поскольку молекулы поверхностного слоя жидкости взаимодействуют не только между собой, но и с молекулами контактирующей с жидкостью среды, то свойства этой среды неизбежно влияют на величину поверхностного натяжения жидкости.
а. Взаимодействие жидкостей с газами очень слабое. Состав газов над жидкостью мало влияет на величину s. Опыты показывают, что величина s для данной жидкости максимальна на границе жидкость – собственный пар (Таблица 18).
| Поверхностное натяжение на границе с газами и жидкостями Таблица 18 | |||||||
| Вещество | Пограничная среда | T, K | s, Н/м | Вещество | Пограничная среда | T, K | s, Н/м |
| Ртуть Hg Ртуть Hg Ртуть Hg Ртуть Hg | Пары Hg Воздух Вода жидк. Спирт жидк. | 290 290 290 290 | 0,490 0,472 0,427 0,399 | Вода H2O Вода H2O Вода H2O Вода H2O | Пары H2O Воздух Бензол жидк. Эфир жидк. | 290 290 290 290 | 0,073 0,073 0,034 0,012 |

|
|
Рассмотрим каплю какой-либо жидкости, помещенную на поверхность другой жидкости (рис.58).
Полагаем каплю настолько малой, чтобы массовыми силами можно было пренебречь.
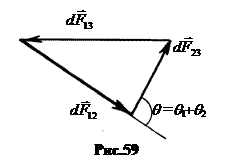 Здесь
Здесь  – силы поверхностного натяжения, действующие на элемент длины dl границы раздела 3-х сред.
– силы поверхностного натяжения, действующие на элемент длины dl границы раздела 3-х сред.
Когда капля примет окончательную форму и будет находиться в равновесии, сумма сил обратится в нуль (рис.59).
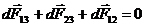 . Так как dF13 = s13dl, dF23 = s23dl, dF12 = s12dl, то, представив dF13 по теореме косинусов и разделив все члены на dl, получаем:
. Так как dF13 = s13dl, dF23 = s23dl, dF12 = s12dl, то, представив dF13 по теореме косинусов и разделив все члены на dl, получаем:
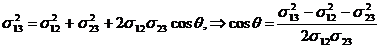 . (17.3)
. (17.3)
Если q ® 0, капля растекается в мономолекулярный слой. В остальных случаях капля растекается до некоторого максимального диаметра и переходит в устойчивое состояние.
Углы q1 и q2, образуемые касательными к поверхностям контактирующих сред, проведенными из границы раздела 3-х сред, называются краевыми углами. Когда q ® 0, говорят о полном смачивании одной жидкостью другой. Например, бензин и вода.
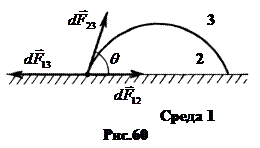
в. Взаимодействие жидкостей с твердыми телами. Если малая капля жидкости находится на плоской поверхности твердого тела (рис.60), то ее равновесию соответствует равенство:  . Спроектировав уравнение на ось x, совпадающую с поверхностью твердого тела, и разделив его на dl, получаем выражение для краевого угла:
. Спроектировав уравнение на ось x, совпадающую с поверхностью твердого тела, и разделив его на dl, получаем выражение для краевого угла: 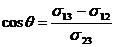 . (17.4)
. (17.4)
 В зависимости от величины q различают:
В зависимости от величины q различают:
При q = 0 – полное смачивание (керосин по стали),

 – частичное смачивание (рис.61-а),
– частичное смачивание (рис.61-а),
 –частичное несмачивание (парафин-вода рис.61-б),
–частичное несмачивание (парафин-вода рис.61-б),
q = p – полное несмачивание (ртуть-стекло).
Взаимодействие молекул жидкости с молекулами твердого тела влияет на форму поверхности жидкости, налитой в сосуд. В результате образуется мениск - поверхность 2-го порядка. Если сосуд широкий, то поверхностные силы уступают массовым, и поверхность мениска совпадает с эквипотенциальной поверхностью силового поля. Если же размеры сосуда малы (@1 мм), то преобладают поверхностные силы. В этом случае мениск имеет самостоятельную кривизну.
4. Зависимость поверхностного натяжения от температуры. С повышением температуры жидкость разрыхляется, расстояние между молекулами увеличивается, а силы притяжения между ними уменьшаются.
С другой стороны, с ростом температуры увеличивается плотность насыщенных паров жидкости, силы притяжения, действующие на молекулы жидкости со стороны молекул пара, увеличиваются.
Оба эти механизма должны приводить к уменьшению поверхностного натяжения с ростом температуры. В критической точке, когда исчезает разница между жидким и газообразным состояниями, коэффициент поверхностного натяжения должен обращаться в нуль.
 Общий вид зависимости s(T) можно найти из термодинамических соображений. Воспользуемся для мысленного эксперимента рамкой с подвижной перекладиной, на которой натянута жидкая пленка (рис.57).
Общий вид зависимости s(T) можно найти из термодинамических соображений. Воспользуемся для мысленного эксперимента рамкой с подвижной перекладиной, на которой натянута жидкая пленка (рис.57).
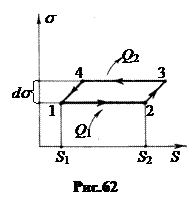
Проведем цикл Карно, растягивая и сжимая пленку. Пусть в состоянии 1 пленка имеет площадь S1 при температуре T. В системе координат s, S этому состоянию соответствует точка 1 (рис.62).
а. Изотермически растянем пленку из состояния 1 в состояние 2, ее площадь увеличится от S1 до S2. Поверхностная энергия при этом также увеличится. Чтобы температура пленки осталась постоянной, к ней надо подвести тепло Q1. Работа расширения A1®2 = s (S2 - S1).
б. Адиабатически растянем пленку из состояния 2 в состояние 3 на бесконечно малую величину площади dS. Температура пленки понизится от T до T - dT, а поверхностное натяжение станет больше на величину ds.
в. Изотермически сожмем пленку, дав возможность ей сократиться при температуре T - dT (переход 3 ® 4). Площадь уменьшится на величину DS = S2 - S1. Для поддержания температуры пленки постоянной, от нее надо отвести тепло Q2.
г. Адиабатически сожмем пленку, дав возможность ей сократиться без притока тепла на величину dS. Температура при этом повысится на величину dT, а поверхностное натяжение уменьшится на величину ds (переход 4 ® 1). Цикл завершен.
Работа этого обратного цикла отрицательна и равна площади бесконечно тонкой трапеции 1 ® 2 ® 3 ® 4. dA = – ds (S2 - S1). От внешних тел получено тепло Q1 - Q2.
КПД цикла  . (17.5)
. (17.5)
Так как цикл идеальный, то по теореме Карно: 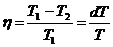 . (17.6)
. (17.6)
Отсюда 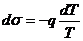 . (17.7)
. (17.7)
Коэффициент q = (Q1 - Q2)| (S2 - S1) есть количество тепла, которое надо подвести к единице площади пленки при совершении цикла. Поскольку пленка совершает обратный цикл, то это тепло в данном случае должно отводиться от пленки.
В общем случае q зависит от температуры и с ростом T несколько уменьшается. Однако во многих случаях величину q можно считать постоянной, что упрощает интегрирование уравнения (17.7). s = – qlnT + lnC. (17.8)
Если постоянную интегрирования lnC найти из условия, что поверхностное натяжение в критической точке исчезает,  , то lnC = qlnTк, и
, то lnC = qlnTк, и
 . (17.9)
. (17.9)
Поскольку T всегда меньше Tк, то выражение (Tк- T) | Tк< 1. Это позволяет разложить логарифм в ряд и ограничиться первым членом, ln(1+x)|x<1= x.  . (17.10)
. (17.10)
На практике коэффициент q определяют, измерив s при некоторой определенной температуре. Удобно взять для этого точку замерзания-плавления Tпл.
Тогда  . (17.11)
. (17.11)
Здесь sпл – поверхностное натяжение в точке замерзания жидкости.
Дата добавления: 2018-04-15; просмотров: 3003; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!