Существуют различные методы защиты от коррозии.
I закон термодинамики -тепло, приложенное к системе, расходуется на изменение внутренней энергии веществ и совершение работы: Q = ΔU + A , где Q -количество тепла , ΔU -внутренняя энергия, общий запас энергии системы (энергия поступательного и вращательного теплового движения молекул, колебательного движения атомов, энергия вращения электронов). Внутренняя энергия – полная энергия системы без потенциальной энергии, обусловленной положением системы в пространстве и без кинетической энергии системы как целого. Количество выделенной или поглощенной теплоты называется тепловым эффектом реакции. В изобарно-изотермических системах при протекании процесса в открытых сосудах при атмосферном давлении (P = const) все тепло, приложенное к системе, идет как на изменение внутренней энергии, так и на совершение работы. Функция, учитывающая эти две составляющие, называется энтальпией или внутренним теплосодержанием системы. H = U+PV, а тепловой эффект при P = const Qv = ΔU+ PΔV=ΔH. ΔH -энтальпия - тепловой эффект реакции при P = const . Размерность кДж/моль, Дж/моль, ккал/моль, кал/моль (1кал = 4,1840 Дж). Для простых веществ энтальпия образования и внутренняя энергия приняты равными нулю. ΔH01 ΔH02 ΔH03 ΔH04 ΔH05 ΔH06 ΔH07 A F B D C E Химические уравнения, в которых указаны тепловые эффекты реакций, называются термохимическими уравнениями. В термохимическом уравнении тепловой эффект приводится на один моль продукта, указывается агрегатное состояние «к», «ж», «г», «т», например: ½ H2 (г) + ½ Сl2 (г)= HCl(г) + 92 кДж , ΔH = – 92 кДж Закон Гесса:Тепловой эффект реакции не зависит от пути перехода, а зависит только от начального и конечного состояния участвующих в реакции веществ. Согласно закону Гесса: ΔH1 = ΔH2 + ΔH3 + ΔH4 = ΔH5 + ΔH6 + ΔH7 Закон позволяет рассчитать тепловой эффект любой промежуточной стадии процесса, которую нельзя осуществить экспериментально. Следствие из закона Гесса: 1)Стандартный тепловой эффект реакции равен сумме стандартных теплот образования продуктов реакции за вычетом суммы стандартных теплот образования исходных веществ. Например, для реакции: nA + mB + … D pC + qD + … ΔH0х.р.= ( pΔH0C + qΔH0D + …) – ( nΔH0A + mΔH0B +…) 2)Тепловой эффект реакции равен сумме теплот сгорания исходных веществ за вычетом суммы теплот сгорания продуктов реакции. Принцип Бертло.Любая система стремится к минимуму энергии, поэтому реакции протекают самопроизвольно при выделении тепла. Однако, при высоких температурах вопреки принципу Бертло, происходят процессы, сопровождающиеся поглощением тепла. 2SO2 +O2 « 2SO3 + 192,5 кДж (ΔH= -192,5 кДж) - экзотермическая химическая реакция - CH4 + CO2 « 2CO + 2H2 – 259,4 кДж (ΔH= +259,4 кДж) - эндотермическая химическая реакция Изобарно-изотермический потенциал или свободная энергия Гиббса. Все системы самопроизвольно стремятся к минимуму энергии (ΔH) , но одновременно к максимуму неупорядоченности (S). Функцией состояния, одновременно отражающей влияние обеих тенденций на направление протекания химических процессов, служит изобарно-изотермический потенциал или свободная энергия Гиббса. Изменение изобарно-изотермического потенциала или свободной энергии Гиббса в химическом процессе: ΔG = ΔH – TΔS (при P =const) Процессы, протекающие в закрытых сосудах при постоянном объеме, характеризуются изменением свободной энергии Гельмгольца ΔF: ΔF = ΔU – TΔS (при V = const) Энергии Гиббса и Гельмгольца являются функциями состояния, т.е. их изменение (ΔG и ΔF) зависят только от начального и конечного состояний и не зависят от пути осуществления процесса. ΔG0р = ∑ΔG0продуктов - ∑ΔG0исходных ΔG и ΔF имеют ту же размерность, что и энтальпия. ΔG0 образования простых веществ принимают равными нулю. ΔGр = ΔHр – TΔSр При низких температурах значение TΔS мало, преобладает энтальпийный фактор(ΔH) и принцип Бертло справедлив - тепловой эффект определяет возможность самопроизвольного протекания реакции. При высоких температурах преобладает энтропийный фактор (– TΔS) и принцип Бертло неприменим. Если система находится в состоянии равновесия, в системе не происходит ни энергетических изменений (ΔH = 0), ни изменений в степени беспорядка (ΔS = 0), то есть ΔG = 0. При постоянстве температуры и давления химические реакции могут самопроизвольно протекать только в таком направлении, при котором энергия Гиббса системы уменьшается (ΔG < 0). Скорость химической реакции –характеризуется изменением концентрации реагирующих веществ за единицу времени. Зависит от: Природы вещества, концентрации реагентов, температуры, катализатора, Закон скорости реакций: aA+bB+…=cC+dD…(продукты реакции) Скорость реакции: , где скорость реакции прямо пропорционально произведению концентраций. a и b порядок реакции реагентов (а+b+…=n)-общий порядок реакции, k-константа скорости реакции(от природы реагентов, от температуры)-k-это скорость реакции при единичной концентрации. Энергия активации - Ea (Дж/моль)— минимальное количество энергии, которое требуется сообщить системе, чтобы произошла реакция. Зависимость скорости реакции от температуры выражает закон Вант-Гоффа: при повышении температуры на каждые 10 градусов скорость гомогенной реакции возрастает на 2-4 раза. , где g - температурный коэффициент, равный .Чтобы произошла реакция, сталкивающиеся частицы должны обладать энергией достаточной для преодоления сил отталкивания, т.е. энергетического барьера (энергия активации Eaкт). При повышении температуры энергия вещества возрастает, возрастает и скорость реакции, что описывается с помощью уравнения Аррениуса. , где A – предэкспоненциальный множитель, R – универсальная газовая постоянная, T – температура, K, Еакт – энергия активации. В логарифмической форме уравнение Аррениуса имеет вид: . Катализаторами называют вещества, которые изменяют скорость химической реакции, но в результате реакции не расходуются. Явление катализа распространено в природе (большинство процессов, происходящих в живых организмах, являются каталитическими) и широко используется в технике (в нефтепереработке и нефтехимии, в производстве серной кислоты, аммиака, азотной кислоты и др.). Большая часть всех промышленных реакций — это каталитические. Катализ - сам процесс влияния катализатора на скорость химической реакции. Отрицательные катализаторы(ингибиторы)- вещества, которые понижают скорость химической реакции. Положительные катализаторы- вещества, которые повышают скорость химической реакции. При гомогенном катализе катализатор находится в том же агрегатном состоянии, что и реагенты.(кислоты, соли, основания) H2О2 + I → H2О + IO H2О2 + IO → H2О + О2 + I При гетерогенном катализе катализатор находится в другом агрегатном состоянии, чем реагенты. (переходные металлы и их оксиды) Химическое равновесие - состояние системы, в котором скорость прямой реакции (V1) равна скорости обратной реакции (V2). Реакции, которые могут одновременно протекать в прямом и обратном направлениях, называются обратимыми. Система обратима, когда скорость реакции прямой реакции, равна скорости обратной. . I2 + H2 ⇄2HI u1=k1[I2]·[H2] - прямая реакция u2=k2[HI]2 – обратная реакция u1=u2 – состояние равновесия Закон действующих масс устанавливает соотношение между массами реагирующих веществ в химических реакциях при равновесии, а также зависимость скорости химической реакции от концентрации исходных веществ. Зависимость скорости реакции от концентрации выражает закон действия масс (закон Гульдберга и Вааге): скорость реакции пропорциональна произведению молярных концентраций реагирующих веществ в степенях, соответствующих их стехиометрическим коэффициентам. Для гомогенной реакции вида mA + nB = pC + qD υ = kc × CAm × CBn, В гетерогенных системах концентрация наиболее конденсированной фазы, на поверхности которой происходит реакция, постоянна и в законе действия масс не учитывается. Например, 2С(тв) + O2(г) → 2CO(г); V = kc·CO2.Закон действия масс справедлив для разбавленных растворов и идеальных газов. Для гетерогенных реакций концентрации твердых и жидких веществ в выражение константы равновесия не входят ввиду их постоянства в гетерогенных реакциях (реагентов и продуктов) изменяется только в наименее упорядоченной гомогенной части системы. Для гетерогенных систем «газ + жидкость + твердое тело» это будет газовая фаза (индивидуальное вещество или газовый раствор), а для систем «жидкость + твердое тело» — жидкая фаза (индивидуальное вещество или жидкий раствор). Предполагается также, что остальные гомогенные части этих систем растворами не являются, а состоят из индивидуальных веществ. Смещение химического равновесия Перевод равновесной химической системы из одного состояния равновесия в другое называется смещением (сдвигом) химического равновесия, при изменении температуры, концентрации, давления. Принцип Ле-Шателье: если на систему в состоянии истинного равновесия воздействовать извне, изменяя термодинамические параметры, то равновесие сместится в таком направлении, которое ослабит эффект внешнего воздействия. Способы смещения равновесия: 1. При увеличении концентрации какого-либо из веществ, участвующих в равновесии, равновесие смещается в сторону расходу этого вещества; при уменьшении концентрации какого-либо из веществ равновесие смещается в сторону образования этого вещества. S2+2O2=2SO2 [S],[O]↑ →, [SO2]↑ ← 2. При увеличении давления путем сжатия системы равновесие сдвигается в сторону уменьшения числа молекул газов, т.е. в сторону понижения давления; при уменьшении давления равновесие сдвигается в сторону возрастания числа молекул газов, т.е. в сторону увеличения давления. CaCO3=CaO+CO2 P↑ ←, P↓ → 1моль=1моль+1моль. 3. При повышении температуры равновесие смещается в направлении эндотермической, а при понижении – в направлении экзотермической реакции. CaCO3=CaO+CO2 -Q t↑ →, t↓ ←; N2+3H2↔2NH3 +Q t↑ ←, t↓ → Растворами называют гомогенные однородные системы, состоящие из двух или более компонентов. В зависимости от агрегатного состояния различают газообразные, жидкие и твердые растворы. Растворы состоят из растворенного вещества и растворителя; в общем случае растворителем считают тот компонент, которого больше. Способы выражения концентрации: Массовая доля-отношение массы растворенного вещества к общей массе раствора: для раствора X; массовая доля – величина безразмерная (выражают в долях единицы либо в процентах). Мольная доля- отношение количества растворенного вещества в молях к суммарному количеству всех молей компонентов раствора; N(X)- величина безразмерная или выраженная в процентах. Молярная концентрация раствора (молярность)- количество молей растворенного вещества в 1 л раствора: c(X), выражают в моль/л. Моляльная концентрация (моляльность) m- число молей растворенного вещества, приходящееся на 1 кг растворителя. Эквивалентная (нормальная) концентрация Сэ (молярная концентрация эквивалентов вещества B определяется числом эквивалентов растворенного веществаnэ в единице объема раствора, размерность моль-экв/литр: где nЭ(B) – количество эквивалентов вещества, μЭ – молярная масса эквивалента (эквивалентная масса). Молярная масса эквивалента (эквивалентная масса) соотносится с молярной массой следующим образом: для кислот: ; для оснований: ; для солей и оксидов: , где nH+ – основность кислоты; nOH- – кислотность основания; nMe – число атомов металла в молекуле соли или оксида; BMe – валентность металла. Отсюда следует, что для кислот CЭ = CM . nH+; для оснований CЭ = CM . nOH-; для солей и оксидов CЭ = CM . nMe . BMe. Титр раствора вещества B определяется массой растворенного вещества, содержащегося в 1 мл раствора; TB, г/мл: , г/мл Осмос (от греч. ōsmós - толчок, давление), диффузия[5] вещества, обычно растворителя, через полупроницаемую мембрану, разделяющую раствор и чистый растворитель или два раствора различной концентрации. Полупроницаемая мембрана - перегородка, пропускающая малые молекулы растворителя, но непроницаемая для более крупных (иногда за счет возникновения сольватной оболочки) молекул растворённого вещества. Выравнивание концентраций по обе стороны такой мембраны возможно лишь при односторонней диффузии растворителя, поэтому всегда идёт от чистого растворителя к раствору, или от разбавленного раствора к концентрированному. Осмос, направленный внутрь ограниченного объёма жидкости, называется эндосмосом, наружу - экзосмосом.Разбавленные растворы хорошо подчиняются законам идеальных газов (pV=nRT). Сходство разбавленных растворов с идеальными газами Вант-Гоффа выразил в виде закона: Pосм = CМ·R·T, где СМ − молярная концентрация раствора (число моль вещества на 1 л раствора); R − универсальная газовая постоянная; T − абсолютная температура. Закон Вант-Гоффа: Осмотического давления определяет давление молекул растворенного вещества на полупроницаемую перепонку, отделяющую раствор от чистого растворителя и непроницаемую для растворенного вещества. Здесь подразумевается давление по отношению к чистому растворителю. Если же мембрана разделяет два раствора, то: Pосм = Pосм 1 - Pосм 2 = (См 1 - См 2)RT Таким образом, осмотическое давление идеального раствора равно тому давлению, которое оказывало бы растворенное вещество, если бы оно, находясь в газообразном состоянии при той же температуре, занимало бы тот же объем, который занимает раствор. Первый закон Рауля: Понижение давления пара растворителя над раствором при постоянной температуре пропорционально мольной доле растворенного вещества. , где Δр − понижение давления пара; р − давление пара растворителя над чистым растворителем; n − число молей растворенного вещества; N − число молей растворителя. Отношение n/(n + N) − мольная доля растворенного вещества (χ). Второй закон Рауля Повышение температуры кипения раствора по сравнению с температурой кипения чистого растворителя Испарение жидкости с поверхности может происходить при любой температуре. Если же парообразование происходит не только со свободной поверхности жидкости, но и внутри ее, то такой процесс называют кипением. Если давление насыщенного пара над раствором меньше, чем над растворителем, то, чтобы довести его до атмосферного, требуется более высокая температура. То есть температура кипения раствора выше температуры кипения чистого растворителя: Dtкипения = tкипения раствора - tкипения чистого растворителя = Кэ · Сm, где Кэ – эбуллиоскопическая константа, являющаяся характеристикой растворителя и не зависящая от природы растворенного вещества, Сm - моляльная концентрация раствора[6]. Для расчетов часто используется следующее выражение закона: Второй закон Рауля. Понижение температуры замерзания раствора по сравнению с температурой замерзания чистого растворителя температура замерзания раствора ниже температуры замерзания чистого растворителя: Δtзамерзания = tзамерзания раствора - tзамерзания чистого растворителя = Ккр · Сm где Ккр - криоскопическая константа, являющаяся характеристикой растворителя и не зависящая от природы растворенного вещества, Сm - моляльная концентрация раствора. Для расчетов часто используется следующее выражение закона: Итак, второй закон Рауля является следствием из первого и формулируется следующим образом: повышение температуры начала кипения и понижение температуры начала кристаллизации идеального раствора (по сравнению с растворителем) пропорциональны суммарной моляльной концентрации всех растворенных веществ при условии, что в пар и в твердую фазу переходит только растворитель. Свойства растворов неэлектролитов. Разбавленные растворы неэлектролитов обладают рядом свойств (коллигативные), количественное выражение которых зависит только от числа находящихся в растворе частиц растворенного вещества и от количества растворителя. Некоторые коллигативные свойства растворов используются для определения молекулярной массы растворенного вещества. Зависимость этих свойств от концентрации выражается 4 уравнениями: 1) Понижения давления пара растворителя над раствором. 2) Понижение температуры кристаллизации раствора. 3) Повышение температуры кипения раствора. Осмотическое давление. Сильными электролитами являются многие хорошо растворимые кислоты(серная, хлороводородная, азотная, хлорная, бромоводородная), также ионные соединения( все соли и гидроксиды). Слабые электролиты- это такие кислоты, как сернистая, борная, уксусная, большинство органических кислот и вода.( почти все органические кислоты и вода; некоторые неорганические кислоты: HF, HClO, HClO2, HNO2, HCN, H2S, HBrO, H3PO4, H2SO3 и др.; некоторые малорастворимые гидроксиды металлов: Fe(OH)3, Zn(OH)2 и др.; а также гидроксид аммония NH4OH.) Электролитическая диссоциация. Молекулы электролитов частично или полностью распадаются на ионы: Степень электролитической диссоциации - показывает отношение числа продиссоциированных молекул общему числу молекул. n- число ионов данного сорта, Константа диссоциации – это константа равновесия системы, возникающей в растворе слабого электролита, например: HCN ⇄ H+ + CN- Применив закон действующих масс, получим K зависит от природы реагирующих веществ и от температуры. Взаимосвязь между степенью и константой диссоциации описывается законом разбавления Оствальда: при разбавлении растворов слабых электролитов степень электролитической диссоциации возрастает: α ≈ Закон разбавления Оствальда- — соотношение, выражающее зависимость эквивалентной электропроводности разбавленного раствора бинарного слабого электролита от концентрации раствора: Свойства электролитов определяются характером ион-ионных и ион-молекулярных взаимодействий, а также изменением свойств и структуры растворителя под влиянием растворенных частиц электролитов. Изотонический коэффициент представляет собой меру увеличения числа частиц в растворе электролита за счет диссоциации. Кроме того он показывает, во сколько раз экспериментально измеренные величины для электролитов, больше теоретически рассчитанных по законам для неэлектролитов: Для того, чтобы математическое выражение законов Вант-Гоффа и Рауля были справедливыми и для разбавленных растворов электролитов, вводится поправочный коэффициент i – изотонический коэффициент или коэффициент Вант-Гоффа. Для растворов электролитов формулы имеют следующий вид: ΔР = ; Δtкип. = iKэб.Сm и Δtзам = iKкр.Сm; Росм. = iСмRТ. Изотонический коэффициент i связан со степенью диссоциации (α) и количества ионов, образующихся при диссоциации одной молекулы электролита (k): i = 1 + α (k –1) или α = (i –1)/(k – 1) Например, для сульфата алюминия: Al2(SO4)3 = 2 Al3+ + 3 SO42-; k = 2 + 3 =5, и при α = 1 i= 5 Ионное произведение водыВода – слабый электролит, диссоциирующий, хотя и весьма незначительно, по уравнению: H2O ⇄ H+ + OH– ; Произведение концентраций ионов H+ и OH– называется ионным произведением воды и является величиной постоянной для водных растворов при н.у.: Кв = [H+]·[ OH- ] = 10-14. Водородный показатель ( pH )При равенстве концентраций [H+] и [ OH- ] в нейтральной среде [H+] = [ OH- ] = 10–7 В кислой среде концентрация концентрация [H+] больше, чем 10–7 моль/л , а в щелочной среде –– меньше, чем 10–7. Кислотность среды выражается величиной водородного показателя рН, рассчитанной по формуле: рН = – lg[H+] 10-1… 10-5 10-6 10-7 10-8 10-9… 10-14 кислая ← [H+] → щелочная нейтральная Щелочность среды выражается величиной рОН, значение которой рассчитывается по формуле рОН = – lg[ОH–]. В кислой среде рН< 7, рОН>7, в щелочной среде рН>7, рОН< 7. Зная рН, легко вычислить рОН, поскольку рН + рОН = 14. Произведение растворимости ( ПР) Абсолютно нерастворимых осадков не существует, любой осадок хоть незначительно, но растворяется, а растворенная часть полностью диссоциирована на ионы: AgClтв⇄ AgCl(раствор) = Ag+ + Cl–; Между осадком и ионами в растворе устанавливается равновесие: AgClтв⇄ Ag+ + Cl–, которое количественно описывается величиной константы равновесия, называемой произведением растворимости: ПР = [Ag+]·[Cl–] Произведение растворимости – произведение концентраций ионов в насыщенном растворе трудно-растворимых веществ при данной температуре. Это величина постоянная и зависит только от температуры. Например, для Ag2S↓⇄2Ag+ + S2– , ПР = [Ag+]2·[ S2–]; для Ag3PO4↓⇄ 3Ag+ + PO43–, ПР = [Ag+]3·[ PO43–]. Сильные электролиты- химическое соединение, молекулы которых в разбавленных растворах практически полностью диссоциированы на ионы. Количественные расчеты характеристик растворов сильных электролитов осуществляют с помощью понятий активности электролита и активностей катионов и анионов и . Которые равны произведению коэффициента активности на концентрацию. Под активностью иона понимают ту эффективную («кажущуюся») концентрацию его, которая соответствует определенным свойствам раствора. Активность иона (а) пропорциональна его концентрации: аиона = f Cиона, где f – коэффициент активности. В водных растворах коэффициент активности зависит от концентрации и валентности всех присутствующих ионов. Для характеристики этой зависимости введено понятие ионная сила раствора (μ). Ионная сила раствора равна полусумме произведения концентраций всех присутствующих в растворе ионов на квадраты их зарядов: Чем больше ионная сила раствора, тем меньше коэффициент активности каждого иона и меньше его активная концентрация. Коэффициент активности находят по табличным данным или по формуле Дебая-Гюккеля: lg f = . Для разбавленных растворов (0,01–0,05н) – по упрощенной формуле: lg f = – 0,5 . Гидролиз - это процесс обменного взаимодействия вещества с водой протекающий с образованием слабодиссоциирующих, газообразных или труднорастворимых веществ. Для многозарядных анионов гидролиз протекает ступенчато,например в случае карбонатов: I ступень K2CO3 + HOH ⇄ KOH + KHCO3 2K+ + CO32– + HOH ⇄ 2K+ + HCO3–+ OH– CO32– + HOH ⇄ HCO3– + OH– II ступень KHCO3 + HOH ⇄ KOH + H2CO3 K+ + HCO3– + HOH ⇄ K+ + H2CO3 + OH– HCO3– + HOH ⇄ Н2CO3 + OH– На первой ступени гидролиза образуются кислые анионы HCO3–, для которых Kд(2) = 4,8∙10-11, а по второй - молекулы H2CO3, легче распадающиеся на ионы (Kд(1) = 4,5∙10-7). Видно, что гидролиз солей по второй ступени проходит в меньшей степени, чем по первой. Таким образом, при стандартных условиях и умеренном разбавлении раствора гидролиз солей, образованных слабой многоосновной кислотой и сильным основанием, протекает преимущественно по первой ступени и приводит к образованию кислых солей.Реакция среды в растворе – щелочная, рН > 7. Для многозарядных катионовгидролиз протекает ступенчато: I ступень Al2(SO4)3 + 2HOH ⇄ 2AlOHSO4 + H2SO4 2Al3+ + 3SO42- + 2HOH ⇄ 2AlOH2+ + 2H+ + 3SO42- Al3+ + HOH ⇄ AlOH2+ + H+ II ступень 2AlOHSO4 + 2HOH ⇄ (Al(OH)2)2SO4 + H2SO4 2AlOH2+ + 2SO42- + 2HOH⇄2Al(OH)2+ + 2SO42- + 2H+ AlOH2+ + HOH ⇄ Al(OH)2+ + H+ III ступень (практически не протекает) (Al(OH)2)2SO4 + 2HOH ⇄ 2Al(OH)3 + H2SO4 2Al(OH)2+ + SO42- + 2HOH ⇄ 2Al(OH)3 + 2H+ + SO42- Al(OH)2+ + HOH ⇄ Al(OH)3↓ + H+ Вторая и третья ступени при обычных условиях практически не реализуются. В результате гидролиза солей слабого многокислотного основания и сильной кислоты образуются основные соли. Реакция среды кислая рН< 7. Равновесное состояние гидролиза характеризуется постоянной величиной (при определенной температуре), называемой константой гидролиза Kг. Для гидролиза по катиону и аниону одновременно: . Kг зависит от константы диссоциации Kдисс. Для солей, образованных слабым основанием можно записать: ; Если соль сильный электролит, то [ион соли] = Cсоли. Тогда .Тогда . Принимая Kв = 10-14 и учитывая pH = -lg[H+]; pKд = -lgKд. Для солей, гидролизующихся по катиону имеем: Для солей, образованных слабой кислотой и гидролизующихся по аниону, проводя аналогичные преобразования, получим: . При гидролизе солей третьего типа (по катиону и аниону) pH зависит только от величин констант диссоциации образующихся слабого основания и слабой кислоты. Так как , то Интенсивность протекания гидролиза характеризуется степенью гидролиза hгидр - отношением числа гидролизованных молекул к общему числу растворённых молекул (выражается в процентах):
Полный необратимый гидролиз
Если основание и кислота, образующие соль, являются не только слабыми электролитами, но и малорастворимы или летучи, или неустойчивы и разлагаются с образованием летучих продуктов (Cr2S3, Al2S3, Cr2(CO3)3, Fe2(CO3)3, Al2(CO3)3), то в этом случае гидролиз соли протекает необратимо:Al2S3 + 6H2O ⇄ 2Al(OH)3↓ + 3H2S↑ Поэтому сульфид алюминия не может существовать в водных растворах, может быть получен только "сухим способом", например, из простых веществ при высокой температуре: 2Al + 3S  Al2S3 и должен храниться в герметичных сосудах, исключающих попадание влаги. Необратимомугидролизуподвергаются соли трехвалентных катионов Al3+, Cr3+ и Fe3+ в присутствии карбонатов и сульфидов щелочных металлов:
Al2S3 и должен храниться в герметичных сосудах, исключающих попадание влаги. Необратимомугидролизуподвергаются соли трехвалентных катионов Al3+, Cr3+ и Fe3+ в присутствии карбонатов и сульфидов щелочных металлов:
2AlCl3 + 3Na2CO3 + 3H2O → 2Al(OH)3↓ + 3CO2↑ + 6NaCl
Cr2(SO4)3 + 3Na2S + 6H2O → 2Cr(OH)3↓ + 3H2S↑ + 3Na2SO4
Особые случаи гидролиза:
а) соли двухвалентных катионов (кроме Ca2+, Sr2+, Ba2+) в присутствии карбонатов натрия или калия гидролизуются с образованием осадков мало растворимых основных карбонатов и углекислого газа: 2Cu(NO3)2 + 2Na2CO3 + H2O ⇄ (CuOH)2CO3↓ + 4NaNO3 + CO2↑
б) гидролиз солей висмута и сурьмы протекает в две ступени с образованием трудно растворимых солей с катионами типа ВiO+ (висмутил), SbO+(антимонил): SbCl3 + H2O → SbOCl↓ + 2HCl Механизм процесса можно представить следующим образом:
I-я ступень: SbCl3 + HOH ⇄ SbOHCl2 + HCl
II-я ступень: SbOHCl2 + HOH ⇄ Sb(OH)2Cl¯ + HCl,
полученная на второй ступени основная (гидроксо-) соль разлагается с выделением воды и образованием оксосоли: Sb(OH)2Cl ®SbOCl + H2O. Аналогичные процессы протекают при гидролизе солей висмута.
в) гидролиз силикатов щелочных металлов протекает с образованием полисиликатов:
I-я стадия 2Na2SiO3 + 2HOH ⇄ 2NaHSiO3 + 2NaOH
2SiO32- + 2HOH ⇄ 2HSiO3- + 2OH-
II-я стадия 2HSiO3- ⇄ Si2O52- + H2O
Суммарно: 2Na2SiO3 + HOH ⇄ Na2Si2O5 + 2NaOH
Реакции, протекающие с изменением степени окисления элементов, относятся к окислительно-восстановительным (О В Р ) реакциям. BaCl2 + Na2SO4 = BaSO4↓ + 2 NaCl - реакция обмена Sn+2Cl2 + Fe+3Cl3 → Fe+2Cl2 + Sn+4Cl4 - ОВР
Уравнение Нернста. Рассмотрим в общем виде процесс, происходящий в окислительно-восстановительном электроде на границе металл – раствор. Электродный потенциал на таком электроде возникает за счет следующей реакции на поверхности электрода:  где
где  - окисленная форма,
- окисленная форма,  - восстановленная форма вещества,
- восстановленная форма вещества,  - число электронов, участвующих в процессе. Для температуры
- число электронов, участвующих в процессе. Для температуры  К (250 С) уравнение Нернста после несложных преобразований принимает вид
К (250 С) уравнение Нернста после несложных преобразований принимает вид 
Направление окислительно-восстановительных реакций. Изменение свободной энергии Гиббса для процесса восстановления  может быть определено по формуле
может быть определено по формуле  Таким образом, чем больше величина электродного потенциала, тем меньше
Таким образом, чем больше величина электродного потенциала, тем меньше  для реакции и, следовательно, тем более сильным окислителем является окисленная форма вещества ( ) и тем более слабым восстановителем – форма восстановленная ( ). Любую окислительно-восстановительную реакцию в общем случае можно представить в виде следующего уравнения
для реакции и, следовательно, тем более сильным окислителем является окисленная форма вещества ( ) и тем более слабым восстановителем – форма восстановленная ( ). Любую окислительно-восстановительную реакцию в общем случае можно представить в виде следующего уравнения  где
где  - окислитель,
- окислитель,  - продукт восстановления окислителя,
- продукт восстановления окислителя,  - восстановитель,
- восстановитель,  - продукт его окисления. Изменение свободной энергии Гиббса для этой реакции запишется как
- продукт его окисления. Изменение свободной энергии Гиббса для этой реакции запишется как  Реакция будет протекать слева направо, если
Реакция будет протекать слева направо, если  , и, следовательно
, и, следовательно  Это выражение представляет собой основной термодинамический критерий, позволяющий определить направление протекания ОВР. Сопоставление величин электродных потенциалов компонентов, участвующих в ОВР, позволяет не только определить возможность ее протекания, но и предсказать вероятные продукты реакции. Рассмотрим для примера следующую реакцию
Это выражение представляет собой основной термодинамический критерий, позволяющий определить направление протекания ОВР. Сопоставление величин электродных потенциалов компонентов, участвующих в ОВР, позволяет не только определить возможность ее протекания, но и предсказать вероятные продукты реакции. Рассмотрим для примера следующую реакцию  . Запишем ее в ионном виде
. Запишем ее в ионном виде  и выпишем все присутствующие в системе окислители и восстановители и, используя таблицу значений стандартных электродных потенциалов (табл.), возможные продукты их восстановления и окисления и соответствующие им величины стандартных электродных потенциалов. Окислителем в реакции будет вещество, имеющее максимальный электродный потенциал. Поэтому в данной реакции окислителем будет
и выпишем все присутствующие в системе окислители и восстановители и, используя таблицу значений стандартных электродных потенциалов (табл.), возможные продукты их восстановления и окисления и соответствующие им величины стандартных электродных потенциалов. Окислителем в реакции будет вещество, имеющее максимальный электродный потенциал. Поэтому в данной реакции окислителем будет  , а продуктом его восстановления -
, а продуктом его восстановления -  (
(  В). Восстановителем будет вещество из приведенного списка с минимальной величиной электродного потенциала – анион
В). Восстановителем будет вещество из приведенного списка с минимальной величиной электродного потенциала – анион  . Соответственно продуктом его окисления -
. Соответственно продуктом его окисления -  (
(  В). Так как электродный потенциал окислителя больше электродного потенциала восстановителя, протекание этой реакции слева направо оказывается возможным. Окончательно, реакция запишется в виде
В). Так как электродный потенциал окислителя больше электродного потенциала восстановителя, протекание этой реакции слева направо оказывается возможным. Окончательно, реакция запишется в виде  Или
Или 
Степень окисления и валентность.Под степенью окисления понимают условный заряд атома, который мог бы возникнуть, если бы все связи в молекуле были ионными. Степень окисления вычисляют исходя из электронейтральности молекул и учитывая, что водород в большинстве соединений (за исключением соединений с металлами) имеет степень окисления +1, кислород –2 (за исключением соединения с фтором, и пероксидных соединений), щелочные металлы +1, щелочноземельные +2, степень окисления в простых веществах равна 0. Например:Na2S+4O3, Na2S+6O4, KMn+7O4, K2Cr+62O7, K2Mn+6O4, Cl02; Степень окисления и валентность могут не совпадать. Наиболее характерно это для органических соединений:
C-4H4 C–2H3OH HC0HO C+4O2 HC+2OOH,
Восстановители. Мерой восстановительной способности является величина ионизационного потенциала.
Восстановителями могут быть:
а) металлы: Ме - nē = Меn+, например Cu - ē = Cu+; Pb - 2ē = Pb2+.
б) отрицательно заряженные ионы неметаллов: 2I‾ - 2ē = I2
S2‾, Se2‾, Te2‾, I‾, Br‾, Cl‾
4) сложные ионы или молекулы, содержащие атомы в промежуточной степени окисления.
Например, Fe2+ - ē = Fe3+ Pb2+ -2ē = Pb4+ Sn2+ - 2ē =Sn4+
SO32‾, CrO2‾, AsO33‾, NO2‾ - ионы,
SO2, NO, MnO2 - молекулы.
Окислители.Мерой окислительной способности являются величины сродства к электрону и электроотрицательности.
Окислителями могут быть:
1) нейтральные атомы неметаллов(на внешнем уровне 4,5,6,7ē ):
Cl2 + 2ē =2Cl‾, S + 2ē = S2‾. Самым сильным окислителем является фтор F2
2) Ионы и молекулы, содержащие атомы металлов и неметаллов в высшей и промежуточной степенях окисления:
Pb4+, Sn4+, Cr3+, Fe3+, (ионы благородных металлов Ag+, Au+ )
CrO42‾, Cr2O72‾, MnO4‾, NO3‾, SO42‾, SeO42‾, ClO4‾.
Классификация ОВР.
1)Реакции межмолекулярного окисления-восстановления(окислитель и восстановитель - в разных молекулах):
- реакции вытеснения Zn + CuSO4 = ZnSO4 + Cu
- реакции синтеза H2 + Br2 = 2HBr
- реакции с участием среды (примеры с KMnO4, K2Сr2O7).
Пример межмолекулярной ОВР. Взаимодействие K2Cr2O7 c KI в кислой среде. Бихромат-ион в кислой среде переходит в Cr3+:
Cr2O72 ‾=2Cr3+
K2Cr2O7 + KI + H2SO4 = Cr2(SO4)3 + I2 + K2SO4 + H2O
1│Cr2O72‾ + 14Н+ +6ē = 2Cr3+ + 7Н2О
3│2I– – 2ē = I2
Cr2O72‾ + 14Н+ + 6I– = 2Cr3+ + 7Н2О + 3I2
2K+ 7SO42– 6K+ = 3SO42– 4SO42– 8K+
K2Cr2O7 + 6KI + 7H2SO4 = Cr2(SO4)3 + 3I2 + 4K2SO4 + 7H2O
2) Реакции диспропорционирования (самоокисления - самовосстановления):
а)H2O2  O2 + 2H2O │H2O2 + 2ē = 2OH‾
O2 + 2H2O │H2O2 + 2ē = 2OH‾
│H2O2 - 2ē = O2↑ + 2H+
б) 3Se + 6KOH = K2SeO3 + 2K2Se + 3H2O
1│Se + 6OH– – 4ē = SeO32– + 3H2O
2│Se + 2ē = Se2–
Участие пероксида водорода в ОВР.
Поскольку в молекулах H2O2 кислород находится в промежуточной степени окисления, пероксид водорода может выполнять
двойственную функцию: и окислителя и восстановителя.
а)H2O2 - окислитель:
2CrCl3 + 3H2O2 + 10NaOH = 2Na2CrO4 + H2O + 6NaCl
2│Cr3+ + 8OH– –3ē = CrO42– + 4H2O
3│H2O2 + 2ē = 2OH‾
б) H2O2 - восстановитель:
2KMnO4 + 5H2O2 + 3H2SO4 = 2MnSO4 + 5O2 + K2SO4 +8H2O
2│MnO4‾ + 8H+ +5ē =Mn2+ + 4H2O
5│H2O2 - 2ē = O2↑ + 2H+
3) Реакции внутримолекулярного окисления-восстановления(окислитель и восстановитель находятся в одной молекуле):
CuI2 = CuI↓ + I2 ↑
Cu2+ + 2I‾ = CuI↓ + I2
2│Cu2++I‾+1ē = CuI ↓
1│ 2I‾ - 2ē = I2
2Cu2+ + 2I‾ = 2CuI↓ + I2
(NH4)2Cr2O7 = Cr2O3 + N2 + 4H2O
2NH4+ + 2Cr2O72‾ = Cr2O3 + N2 + 4H2O
1│Cr2O72‾ + 4HOH - 6ē = Cr2O3 + 8OH‾
1│2NH4+ + 6ē = N2 +8H+
Cr2O72‾ + 4H2O +2NH4+ = Cr2O3+8H2O + N2
Двойной электрический слой – представляет собой две области объемного заряда разного знака, расположенные по обе стороны границы раздела фаз. Условиями для образования двойного электрического слоя являются: - контакт двух разнородных фаз; - некомпенсированный перенос заряда через поверхность раздела фаз.Переход ионов из одной фазы в другую, либо адсорбция ионов на поверхности одной из фаз. Образование двойного электрического слоя, таким образом, обычно происходит при контакте твердой и жидкой фаз.


 Рис. 2а. на границе активный металл – раствор. Рис. 2б. на границе неактивный металл – раствор; Например, при контакте активного металла с жидкой фазой (раствором) (рис.2а) свободная энергия катионов в металле обычно выше, чем в жидкости, поэтому начинается переход этих ионов с поверхности металла в жидкость с образованием в приповерхностном слое жидкости области положительного объемного заряда. Соответственно в приповерхностной области металла возникает область отрицательного объемного заряда, обусловленного избытком электронов. По мере увеличения концентрации катионов в растворе усиливается напряженность электрического поля в двойном электрическом слое, которое тормозит переход ионов с поверхности металла в раствор и наоборот ускоряет их адсорбцию на поверхности металла. Таким образом, при определенной напряженности электрического поля в двойном электрическом слое устанавливается динамическое равновесие между количеством ионов поступающих в раствор и возвращающихся обратно на поверхность металла. В случае неактивного металла (рис.2б) в первый момент преобладает процесс адсорбции катионов, вследствие чего на металле образуется область объемного положительного заряда, а избыток противоионов формирует со стороны жидкости вблизи поверхности металла область отрицательного объемного заряда. В конечном итоге, как и в первом случае, устанавливается динамическое равновесие, отвечающее определенной напряженности электрического поля в двойном электрическом слое. По такому механизму двойной электрический слой может формироваться за счет адсорбции как катионов, так и анионов, причем по своей химической природе они могут отличаться от состава твердой фазы; Образование двойного электрического слоя сопровождается появлением межфазной разности потенциалов, которая в электрохимии называется электродным потенциалом.
Рис. 2а. на границе активный металл – раствор. Рис. 2б. на границе неактивный металл – раствор; Например, при контакте активного металла с жидкой фазой (раствором) (рис.2а) свободная энергия катионов в металле обычно выше, чем в жидкости, поэтому начинается переход этих ионов с поверхности металла в жидкость с образованием в приповерхностном слое жидкости области положительного объемного заряда. Соответственно в приповерхностной области металла возникает область отрицательного объемного заряда, обусловленного избытком электронов. По мере увеличения концентрации катионов в растворе усиливается напряженность электрического поля в двойном электрическом слое, которое тормозит переход ионов с поверхности металла в раствор и наоборот ускоряет их адсорбцию на поверхности металла. Таким образом, при определенной напряженности электрического поля в двойном электрическом слое устанавливается динамическое равновесие между количеством ионов поступающих в раствор и возвращающихся обратно на поверхность металла. В случае неактивного металла (рис.2б) в первый момент преобладает процесс адсорбции катионов, вследствие чего на металле образуется область объемного положительного заряда, а избыток противоионов формирует со стороны жидкости вблизи поверхности металла область отрицательного объемного заряда. В конечном итоге, как и в первом случае, устанавливается динамическое равновесие, отвечающее определенной напряженности электрического поля в двойном электрическом слое. По такому механизму двойной электрический слой может формироваться за счет адсорбции как катионов, так и анионов, причем по своей химической природе они могут отличаться от состава твердой фазы; Образование двойного электрического слоя сопровождается появлением межфазной разности потенциалов, которая в электрохимии называется электродным потенциалом.  электродный потенциал- разность потенциалов, которая возникает на границе раздела металл-раствор. Если концентрация ионов в растворе равна 1 моль/л, такой потенциал стандартный
электродный потенциал- разность потенциалов, которая возникает на границе раздела металл-раствор. Если концентрация ионов в растворе равна 1 моль/л, такой потенциал стандартный  стандартный электродный потенциал
стандартный электродный потенциал
Водородный электрод представляет собой пару (  ) / 2
) / 2  при концентрации , равной 1г-ион/л и при давлении газообразного водорода 1 атм.
при концентрации , равной 1г-ион/л и при давлении газообразного водорода 1 атм.
Водородный электрод представляет собой платиновую пластину, поверхность которой покрыта слоем мелкодисперсной платины (платиновой «чернью») и которая помещается в раствор серной кислоты Водородный электрод сравнения соединяют при помощи электролитического мостика с электродом, потенциал которого хотят измерить, и тем или иным способом фиксируют величину ЭДС  образовавшегося гальванического элемента
образовавшегося гальванического элемента 
Ряд напряжений металлов.Рассмотрим электроды первого рода вида  . Для такого электрода каждому металлу может быть поставлен в соответствие свой стандартный электродный потенциал. Если теперь расположить все металлы в порядке возрастания стандартных электродных потенциалов, получится ряд напряжений металлов. Ряд начинается литием и заканчивается благородными металлами – платиной, осмием, иридием. Слева в ряду напряжений стоят металлы, являющиеся очень сильными восстановителями. Их стандартный электродный потенциал
. Для такого электрода каждому металлу может быть поставлен в соответствие свой стандартный электродный потенциал. Если теперь расположить все металлы в порядке возрастания стандартных электродных потенциалов, получится ряд напряжений металлов. Ряд начинается литием и заканчивается благородными металлами – платиной, осмием, иридием. Слева в ряду напряжений стоят металлы, являющиеся очень сильными восстановителями. Их стандартный электродный потенциал  В. Катионы этих металлов в водных растворах окислительными свойствами не обладают. Металлы, располагающиеся в конце ряда напряжений, представляют собой очень слабые восстановители. Зато их катионы (
В. Катионы этих металлов в водных растворах окислительными свойствами не обладают. Металлы, располагающиеся в конце ряда напряжений, представляют собой очень слабые восстановители. Зато их катионы ( 

 и т.д.) являются сильными окислителями.
и т.д.) являются сильными окислителями.
За исключением лития, для металлов, располагающихся в главных подгруппах таблицы Менделеева, стандартный электродный потенциал уменьшается по подгруппе сверху вниз. Наоборот, для металлов побочных подгрупп стандартный электродный потенциал увеличивается сверху вниз. Указанная закономерность объясняется характером изменения радиуса атома и энергии ионизации в подгруппах.
Металлы, имеющие отрицательное значение стандартного электродного потенциала (стоящие в ряду напряжений левее водорода), вытесняют водород из кислот. Металлы, стандартный электродный потенциал которых меньше -0,413В (стоящие в ряду напряжений левее марганца включительно), способны вытеснять водород из воды. В химических источниках тока и при электрохимической коррозии металл с меньшей величиной электродного потенциала является анодом по отношению к металлу с большей величиной потенциала.
Гальванический элемент- устройство, позволяющее получать электрический ток за счет проведения химической реакции. химический источник электрического тока, названный в честь Луиджи Гальвани. Принцип действия гальванического элемента основан на взаимодействии двух металлов через электролит, приводящем к возникновению в замкнутой цепи электрического тока. ЭДС гальванического элемента зависит от материала электродов и состава электролита.

 Работа химических источников тока (гальванических элементов, аккумуляторов) основана на том, что процессы окисления и восстановления в ОВР пространственно разделены и протекают каждый на поверхности своего электрода. Рассмотрим для примера работу ГЭ Даниэля, схему которого можно представить в виде
Работа химических источников тока (гальванических элементов, аккумуляторов) основана на том, что процессы окисления и восстановления в ОВР пространственно разделены и протекают каждый на поверхности своего электрода. Рассмотрим для примера работу ГЭ Даниэля, схему которого можно представить в виде 
На границе раздела цинк – раствор протекает процесс  а на границе медь – раствор –
а на границе медь – раствор –  которые сопровождаются появлением соответствующих электродных потенциалов на каждом из электродов.
которые сопровождаются появлением соответствующих электродных потенциалов на каждом из электродов.
Таким образом, цинк заряжается отрицательно, а медь - положительно относительно раствора. Если внешняя цепь разомкнута (цинк и медь не соединены проводником), в системах быстро устанавливается равновесие. Если теперь электроды замкнуть, то электроны по внешней цепи будут переходить с цинка на медь. Тем самым равновесие в системах нарушится: на цинковом электроде начнется растворение цинка, а на медном – восстановление катионов меди. Возникающие в результате последнего процесса избыточные анионы  будут перемещаться через электролитический мостик от медного к цинковому электроду. Суммарный процесс выражается следующим уравнением окислительно-восстановительной реакции
будут перемещаться через электролитический мостик от медного к цинковому электроду. Суммарный процесс выражается следующим уравнением окислительно-восстановительной реакции  Таким образом, в рассматриваемом гальваническом элементе цинк является анодом, а медь – катодом.
Таким образом, в рассматриваемом гальваническом элементе цинк является анодом, а медь – катодом.
ЭДС ( ) химического источника тока определяется, как  где
где  ,
,  - равновесный электродный потенциал катода и анода соответственно. Для рассматриваемого случая
- равновесный электродный потенциал катода и анода соответственно. Для рассматриваемого случая 



ЭДС химического источника тока при прочих равных условиях зависит от разности стандартных электродных потенциалов электродов и логарифма отношения концентраций растворов в каждом из электродов. Это позволяет создавать так называемые концентрационные источники тока, в которых ЭДС возникает только за счет разности концентраций одного и того же электролита. Примером такого источника тока является элемент  . В качестве анода в этом случае будет выступать электрод с меньшей концентрацией нитрата серебра, а в качестве катода – с большей. ЭДС рассчитывается по формуле:
. В качестве анода в этом случае будет выступать электрод с меньшей концентрацией нитрата серебра, а в качестве катода – с большей. ЭДС рассчитывается по формуле: 
Уравнение Нернста — уравнение, связывающее окислительно-восстановительный потенциал системы с активностями веществ, входящих в электрохимическое уравнение, и стандартными потенциалами окислительно-восстановительных пар.  где E — электродный потенциал,
где E — электродный потенциал,  — стандартный электродный потенциал, измеряется в вольтах; R — универсальная газовая постоянная, равная 8.31 Дж/(моль·K); T — температура в кельвинах; F — число Фарадея, равное 96485,35 Кл/моль; n — число моль электронов, участвующих в процессе;
— стандартный электродный потенциал, измеряется в вольтах; R — универсальная газовая постоянная, равная 8.31 Дж/(моль·K); T — температура в кельвинах; F — число Фарадея, равное 96485,35 Кл/моль; n — число моль электронов, участвующих в процессе;  и
и  — активности соответственно окисленной и восстановленной форм вещества, участвующего в полуреакции. Для температуры К (250 С) уравнение Нернста: Для электродов обратимого по катиону:
— активности соответственно окисленной и восстановленной форм вещества, участвующего в полуреакции. Для температуры К (250 С) уравнение Нернста: Для электродов обратимого по катиону:  и
и  Для электродов обратимого по аниону:
Для электродов обратимого по аниону: 
Электролиз – процесс, при котором химические реакции протекают под воздействием электрического тока. При электролизе в раствор или расплав электролита помещается система электродов, состоящая из катода и анода, на которую подается постоянное (реже – переменное) напряжение. Катод подсоединяется к отрицательному полюсу источника напряжения, а анод – к положительному. В электрическом поле положительно заряженные ионы – катионы – движутся к катоду, а отрицательно заряженные ионы – анионы – к аноду. 
Электролиз расплавов солей или оксидов используют как метод получения высокоактивных металлов(натрия, алюминия, магния, кальция), легко вступающих во взаимодействие с водой, например: 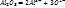 К(-):
К(-):  ; A(+):
; A(+):  -6e=
-6e= 
 ;
;  O2= 2CO;
O2= 2CO;  O2= 2CO2
O2= 2CO2
Электролиз расплавов – очень энергоёмкий метод, т.к соли и оксиды плавятся при высоких температурах. Однако для активных металлов это единственный метод, позволяющий получить их в свободном виде.
В зависимости от инертного электролита электролиз проводится в нейтральной, кислой и щелочной среде. При выборе инертного электролита необходимо учесть, что никогда не восстанавливаются на катоде в водном растворе катионы металлов, являющихся типичными восстановителями (  и никогда не окисляются на аноде кислород анионов оксокислот с элементом в высшей степени окисления
и никогда не окисляются на аноде кислород анионов оксокислот с элементом в высшей степени окисления 
2KCl+2H2O=H2+Cl2+2KOH
K(-):2H+2e=H2
A(+):2Cl-2e=Cl2
Электролиз с растворимым анодом растворяясь отдает (e)
K(-):  +2e=N
+2e=N 
A(+):  - 2e=N
- 2e=N 
Коррозия – это физико-химический процесс взаимодействия металла с окружающей средой, ведущий к разрушению металла. При этом происходит гетерогенное окисление металла, сопровождаемое восстановлением одного или нескольких компонентов среды. Если среда электропроводна, такие реакции имеют электрохимическую природу. Коррозия ускоряется при трении, радиации и высокой скорости потока среды. Различают следующие типы коррозии:
1. Химическая (газовая) коррозия – протекает под действием неэлектролитов.
2. Электрическая коррозия – проявляется при протекании через металл электрического тока. Усиливается при увеличении плотности тока.
3. Электрохимическая коррозия – разрушение металлов под действием растворов электролитов.
4. Биокоррозия – разрушение металлов под действием живых организмов или продуктов их жизнедеятельности.
Химическая коррозия— взаимодействие поверхности металла с коррозионно-активной средой, не сопровождающееся возникновением электрохимических процессов на границе фаз. В этом случае взаимодействия окисление металла и восстановление окислительного компонента коррозионной среды протекают в одном акте. Например, образование окалины при взаимодействии материалов на основе железа при высокой температуре с кислородом: 4Fe + 3O2 → 2Fe2O3
При электрохимической коррозии ионизация атомов металла и восстановление окислительного компонента коррозионной среды протекают не в одном акте и их скорости зависят от электродного потенциала металла (например, ржавление стали в морской воде).
Электрохимическая коррозия- разрушение металла в среде электролита с возникновением внутри системы электрического тока.Важнейший и наиболее распространенный тип коррозии – электрохимическая коррозия. Условием возникновения электрохимической коррозии является контакт двух разнородных металлических фаз при наличии электропроводящей среды, чаще всего – пленки влаги. Причина коррозии – образование микрогальванических пар. Из двух контактирующих фаз та, которая имеет наименьший электродный потенциал, является анодом и окисляется в процессе коррозии. Вторая фаза будет катодом: на ее поверхности будет происходить восстановление компонента среды, который называется деполяризатором. Например, при наличии в месте контакта меди и цинка пленки влаги возникает микрогальванический элемент, схему которого можно записать в виде  . Согласно данным табл.
. Согласно данным табл.  В,
В,  В. Таким образом,
В. Таким образом,  . Поэтому цинк будет анодом, а медь – катодом. Цинк будет окисляться и переходить в раствор
. Поэтому цинк будет анодом, а медь – катодом. Цинк будет окисляться и переходить в раствор 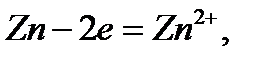 а, на меди будет восстанавливаться деполяризатор – в данном случае – вода
а, на меди будет восстанавливаться деполяризатор – в данном случае – вода 
Различают следующие основные разновидности электрохимической коррозии: контактная коррозия; межкристаллитная коррозия; - коррозия под напряжением.
Существуют различные методы защиты от коррозии.
1. Рациональное конструирование приборов и аппаратуры с тем, чтобы максимально уменьшить число и размеры коррозионно-опасных участков и предусмотреть особые меры защиты таких участков.
2. Замена металлов на коррозионно-стойкие сплавы или неметаллические материалы (пластмассы, керамику, стекло).
3. Термическая обработка металлов с целью повышения однородности структуры металла и снятия механических напряжений (частично устраняет межкристаллитную коррозию и коррозию под напряжением).
4. Изоляция от внешней среды. Для этого используют а) лаки, краски; б) неорганические покрытия (эмали, пленки оксидов, нитридов, фосфатов и т.п.) в) металлические покрытия.
Металлические покрытия разделяют на анодные и катодные. Анодным называется покрытие, металл которого является более активным по отношению к металлу основы. Например, оцинкованное или хромированное железо. При электрохимической коррозии такое покрытие выступает в качестве анода и, разрушаясь, защищает металл основы. Катодным называется покрытие на основе менее активного металла. Например, луженное (покрытое оловом) или никелированное железо. Нарушение такого покрытия усиливает электрохимическую коррозию защищаемого металла.
5. Модификация внешней среды. Состоит в удалении окислителей и введении ингибиторов коррозии в жидкую среду. Используется в основном в технических устройствах, в которых жидкая среда циркулирует по замкнутому контуру, например, в теплообменной аппаратуре.
6. Активные электрохимические методы защиты. Заключаются в создании на защищаемом объекте более отрицательного, чем равновесный, электродного потенциала. Подразделяются на протекторную и катодную защиту. Протекторная защита используется в основном для защиты крупных технических объектов, например, трубопроводов различного назначения. Рядом с объектом закапывают слитки из более активного металла (цинка, алюминия и т.п.) и соединяют их электрически с защищаемым объектом. В этом случае в первую очередь подвергается коррозии более активный металл.
Катодная защита состоит в том, что на защищаемый объект от генератора подается отрицательный относительно земли потенциал.
Химические источники тока, в которых электродные процессы практически обратимы, называются аккумуляторами.
Например, в качестве аккумулятора можно использовать приведенный выше концентрационный элемент. Работу аккумулятора рассмотрим на примере широко используемого на транспорте свинцового аккумулятора.
Свинцовый аккумулятор состоит из решетчатых свинцовых пластин, половина из которых заполнена диоксидом свинца, а другая половина – губчатым свинцом.

Пластины погружены в 35 – 40% раствор серной кислоты. При таком содержании серной кислоты удельная электропроводность электролита максимальна. Металлический свинец служит анодом, а диоксид свинца – катодом. При работе аккумулятора – его разряде – на аноде происходит окисление свинца по реакции
 , а на катоде – восстановление диоксида свинца
, а на катоде – восстановление диоксида свинца 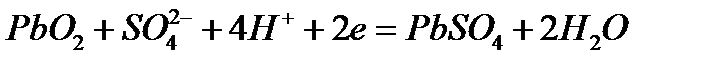 . Во внутренней цепи (в растворе серной кислоты) сульфат-ионы движутся к аноду, а катионы водорода – к катоду.
. Во внутренней цепи (в растворе серной кислоты) сульфат-ионы движутся к аноду, а катионы водорода – к катоду.
Суммарное уравнение окислительно-восстановительной реакции, протекающей при работе свинцового аккумулятора, может быть представлено в виде  .
.
ЭДС полностью заряженного свинцового аккумулятора приблизительно равна 2 В. По мере разряда аккумулятора, расходуются синец, диоксид свинца, а также серная кислота. Для зарядки аккумулятор подключают к внешнему источнику тока плюсом к плюсу, минусом к минусу. В результате, анод аккумулятора при зарядке становится катодом, и на нем идет процесс восстановления  , а катод – анодом, и на нем происходит процесс окисления
, а катод – анодом, и на нем происходит процесс окисления  . Суммарное уравнение окислительно-восстановительного процесса при зарядке аккумулятора имеет вид
. Суммарное уравнение окислительно-восстановительного процесса при зарядке аккумулятора имеет вид  . Таким образом, при зарядке получаются те компоненты, которые необходимы для его работы.
. Таким образом, при зарядке получаются те компоненты, которые необходимы для его работы.
Принцип работы свинцово-кислотных аккумуляторов основан на электрохимических реакциях свинца и диоксида свинца в сернокислотной среде.
Энергия возникает в результате взаимодействия оксида свинца и серной кислоты до сульфата (классическая версия).
Во время разряда происходит восстановление диоксида свинца на катоде и окисление свинца на аноде. При заряде протекают обратные реакции, к которым в конце заряда добавляется реакция электролиза воды, сопровождающаяся выделением кислорода на положительном электроде и водорода — на отрицательном. Химическая реакция (слева-направо — разряд, справа-налево — заряд):
Катод:  Анод:
Анод: 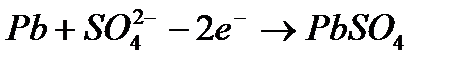 В итоге получается, что при разряде аккумулятора расходуется серная кислота из электролита, плотность электролита падает, а при заряде, серная кислота выделяется в раствор электролита из сульфатов, плотность электролита растёт.
В итоге получается, что при разряде аккумулятора расходуется серная кислота из электролита, плотность электролита падает, а при заряде, серная кислота выделяется в раствор электролита из сульфатов, плотность электролита растёт.
Железо-никелевый аккумулятор Половина реакции на катоде:  ; и на аноде:
; и на аноде:  При разряде реакция протекает слева направо, при заряде справа налево.
При разряде реакция протекает слева направо, при заряде справа налево.
Двойные и комплексные соли. Соединения типа Cu(OH)2; HF; А12(SO4)3; KCI; Fe(CN)2; Ва(NОз)2, часто называют соединениями первого порядка, или валентными соединениями. Соли в растворах диссоциируют с образованием всех ионов, которые содержались в простых солях, т.е. ведут себя как смеси этих солей. Такие соединения называют двойными солями.
Комплексными соединениями называют вещества, содержащие "центральный атом" – комплексообразователь, с которым в неионогенной связи находится определенное количество атомов или молекул, называемых лигандами, составляющих вместе с комплексообразователем внутреннюю сферу.
Теория Вернера: Согласно координационной теории, в молекуле любого элемента комплексного соединения один из ионов, обычно положительно заряженный, занимает центральное место и называется комплексообразователем. Вокруг него координировано некоторое число противоположно заряженных ионов или электро-нейтральных молекул называемых лигандами и образующих внутреннюю координационную сферу соединения. Остальные ионы, не разместившиеся во внутренней сфере, находятся на более далеком расстоянии от центрального иона, составляя внешнюю координационную сферу. Число окружающих центральный ион, называется координационным числом.( Число, показывающее, сколько монодентатных лигандов координирует вокруг себя центральный ион)
Координационное число зависит от: химической природы иона–комплексообразователя([Сu(NНз)4]2+ и [Со(NНз)6]2+), степени окисления комплексообразователя и заряда лигандов, химической природы лиганда([AlI4]‑; [AlF6]3‑)
Устойчивость комплексных соединений- Устойчивость комплексных соединений характеризуется степенью диссоциации комплексных ионов, которая может быть выражена через константу равновесия, называемую константой нестойкости Кн. Чем больше величина этой константы, тем сильнее данный комплекс диссоциирует, тем менее он устойчив. Комплексное соединение [Ag(NH3)2]NО3 диссоциирует по ступеням:
[Ag(NH3)2]NО3 = |Ag(NH3)2]+ + NОз‑ – первая ступень
[Ag(NH3)2]+  Ag+ + 2 NНз0 – вторая ступень.
Ag+ + 2 NНз0 – вторая ступень.
Следует отметить, что диссоциация на ступени I происходит по типу диссоциации сильных электролитов, т.е. почти полностью, а диссоциация по ступени II идет лишь в незначительной степени, т.е. как у слабых электролитов, для которых константа нестойкости определяется по закону действия масс:  Процесс диссоциации и константа нестойкости комплексного иона [Ag(СN)2]‑ выражается следующим образом: [Ag(СN)2]‑ Ag+ + 2СN‑
Процесс диссоциации и константа нестойкости комплексного иона [Ag(СN)2]‑ выражается следующим образом: [Ag(СN)2]‑ Ag+ + 2СN‑ 
При сравнении величин констант нестойкости комплексных ионов видно, что аммиачный комплекс серебра менее прочный, чем цианидный, так как при диссоциации первого в раствор переходит больше свободных ионов, чем при диссоциации второго. Чем меньше Kн, тем более прочен данный комплекс. В структуре комплексного соединения различают координационную (внутреннюю) сферу, состоящую из центральной частицы — комплексообразователя (ион или атом) — и окружающих ее лигандов (ионы противоположного знака или молекулы). Ионы, находящиеся за пределами координационной сферы, образуют внешнюю сферу комплексного соединения. Число лигандов вокруг комплексообразователя называется его координационным числом. Внутренняя сфера (комплекс) может быть анионом, катионом и не иметь заряда. Например, в комплексном соединении K3[Fe(CN)6] внешняя сфера — 3K+, внутренняя сфера [Fe(CN)6]3-, где Fe3+ — комплексообразователь, а 6CN- — лиганды, причем шесть — координационное число. Таким образом, комплексное соединение (как правило) в узлах кристаллической решетки содержит комплекс, способный к самостоятельному существованию и в растворе.
Комплексообразователи. Способность элемента к образованию комплексных соединений относится к важнейшим его химическим свойствам. Она зависит от строения внешнего электронного уровня атома элемента и определяется его положением в периодической системе Д. И. Менделеева. Как правило, комплексообразователями являются атомы или чаще ионы металлов, имеющие достаточное число свободных орбиталей. При образовании химических связей с лигандами комплексообразователи выполняют роль акцепторов. Возможность участия тех или иных орбиталей центрального атома в комплексообразовании меняется от периода к периоду. Способность элементов к комплексообразованию от периода к периоду растет. При последовательном переходе от одного периода к другому растет координационное число элементов.
Лиганды.Лигандами в комплексных соединениях могут служить анионы F-, ОН", CN_, SCN-, N0^, С023 ", С2ОтГ и др.; нейтральные молекулы Н20, NH3, CO, NO, F2, N2H4, NH2—СН2— —СН2—NH2 (этилендиамин) и т. д. Почти все лиганды обладают одной или несколькими неподеленными парами электронов (NH3, Н20, F~, ОН"). Иногда роль лигандов играют молекулы, не содержащие неподеленных пар электронов, но имеющие электроны, участвующие в образовании л-связи. Донорные свойства лигандов реализуются за счет s- и р-атомных орбиталей, а акцепторные— за счет вакантных р- и d-орбиталей.

Теория кристаллического поля
Основные положения:
1. Между комплексообразователем и лигандами существует электростатическое притяжение.
2. При сближении лигандов с комплексообразователем происходит изменение энергетического состояния d- орбиталей, которое зависит от их ориентации в пространстве
Схема энергетических уровней d-орбиталей центрального иона: а- свободный ион, б- ион в гипотетическом сферическом поле, в- ион в октаэдрическом поле лигандов.
Ориентация орбиталей dZ2 (а), dX2-У2(б), dXZ (в) в октаэдрическом поле лигандов (условно изображены в виде шариков)
3.  Величина энергии расщепления ∆ зависит от природы комплексообразователя и лигандов, конфигурации комплекса.
Величина энергии расщепления ∆ зависит от природы комплексообразователя и лигандов, конфигурации комплекса.
4. Чем больше ∆,тем прочнее комплекс.
∆тем больше, чем больше заряд комплексообразователя и чем сильнее поле, создаваемое лигандами. По мере возрастания ∆ лиганды расположены в спектрохимический ряд:
ЭДТА, En, CO, CN-, NH3, CNS-, H2O, OH-, F-, CI-, Br-, I-
Сильное поле среднее слабое поле
Заполнение орбиталей происходит по принципам Паули и наименьшей энергии. Правило Хунда не всегда выполняется. Если ∆ >р, где р- это энергия, затрачиваемая на спаривание электронов, то правило Хунда не выполняется.
Ограниченность теории кристаллического поля: Не объясняет свойства комплексных соединений s- и p- элементов и d-элементов в высших степенях окисления.
Метод молекулярных орбиталей. Рассматривает комплекс как единую квантово-механическую систему, в которой отдельные атомы и молекулы теряют свои индивидуальные черты. Валентные электроны располагаются на многоцентровых МО, охватывающих ядра комплексообразователя и всех лигандов.
Метод Валентных Связей
Согласно теории валентных связей при образовании комплексов возникают донорно–акцепторные связи за счет неподеленных электронных пар лигандов и свободных гибридных орбиталей комплексообразователя. Например, образование комплексных ионов [NH4]+ и [Cu(NHз)4]2+ представляется следующим образом:
Молекулы аммиака являются донорами, так как имеют неподеленную пару электронов, а ионы Сu2+, имеющие свободные орбитали, – акцепторами. Таким образом, при образовании комплексных соединений акцептором является комплексообразователь, а донорами – лиганды.
Структура комплекса определяется в основном электронным строением центрального иона, но она также зависит и от природы лигандов.
Элементарные полупроводники К элементарным полупроводникам относятся либо типичные неметаллы, либо вещества, сочетающие в себе свойства металлов и неметаллов. В настоящее время известно всего 12 элементов, которые в той или иной аллотропной форме способны проявлять полупроводниковые свойства. В таблице Менделеева они располагаются в главных подгруппах и группируются вдоль диагонали, проходящей через бор – йод. Ниже представлена часть таблицы Менделеева, в которой располагаются элементы, проявляющие полупроводниковые свойства, и указана ширина запрещенной зоны (в эВ) для полупроводниковых аллотропных модификаций этих элементов. Для углерода приведена ширина запрещенной зоны для алмаза, для олова - a-модификации.
| периода | Номер группы | ||||
| III | IV | V | VI | VII | |
| 2 | B 1,55 | C 5,7 | |||
| 3 | Si 1,12 | P 1,5 | S 2,5 | ||
| 4 | Ge 0,665 | As 1,2 | Se 1,8 | ||
| 5 | Sn 0,08 | Sb 0,12 | Te 0,36 | I 1,25 | |
Из приведенных в таблице данных можно сделать следующий вывод: ширина запрещенной зоны по периоду возрастает слева направо, а в группах – уменьшается сверху вниз. Уменьшение ширины запрещенной зоны связано в первую очередь с усилением металлических свойств элементов.
Алюминий— элемент главной подгруппы третьей группы третьего периода периодической системы химических элементов Д. И. Менделеева, с атомным номером 13. Обозначается символом Al (лат. Aluminium). Относится к группе лёгких металлов. Наиболее распространённый металл и третий по распространённости химический элемент в земной коре (после кислорода и кремния). Простое вещество алюминий — лёгкий, парамагнитный металл серебристо-белого цвета, легко поддающийся формовке, литью, механической обработке. Алюминий обладает высокой тепло- и электропроводностью, стойкостью к коррозии за счёт быстрого образования прочных оксидная плёнка, защищающих поверхность от дальнейшего взаимодействия.
Химические свойства
При нормальных условиях алюминий покрыт тонкой и прочной оксидной плёнкой и потому не реагирует с классическими окислителями: с H2O (t°);O2, HNO3 (без нагревания). Благодаря этому алюминий практически не подвержен коррозии и потому широко востребован современной индустрией. Однако при разрушении оксидной плёнки (например, при контакте с растворами солей аммония NH4+, горячими щелочами или в результате амальгамирования), алюминий выступает как активный металл-восстановитель. Легко реагирует с простыми веществами. Со щелочами (с образованием тетрагидроксоалюминатов и других алюминатов):
2Al + 2NaOH + 6H2O = 2Na[Al(OH)4] + 3H2
2(NaOH•H2O) + 2Al = 2NaAlO2 + 3H2
Легко растворяется в соляной и разбавленной серной кислотах:
2Al + 6HCl = 2AlCl3 + 3H2
2Al + 3H2SO4(разб) = Al2(SO4)3 + 3H2
Нахождение в природе. Алюминий— самый распространенный металл в природе. Общее содержание алюминия в земной коре составляет 8,8%. В свободном виде алюминия в природе нет. Важнейшие природные соединения (природные руды):
алюмосиликаты — Na2О·Al2О3·2SiО2 и К2О·А12О3·2SiО2;
бокситы — А12О3·nН2О, (AlO(OH))
корунд — А12О3,
криолит — Na3[AlF6] или 3NaF·AlF3. Алюмосиликаты составляют большую часть массы земной коры.
Получение.
1. AlO(OH)®Al(OH)3®А12О3
2. Алюминий получают электролизом оксида алюминия А12О3 в расплаве криолита. Процесс электролиза в конечном итоге сводится к разложению А12О3 электрическим током:
| 2А12О3 | 950°, Na3[AlF6] | 4Al + 3O2↑ |
| Электр.ток |
(-) Катод: 4Al3+ +12ē = 4Al0
(+) Анод: 6 O2- -12ē = 3O2 ↑
Физические свойства. Алюминий — легкий, серебристо-белый, пластичный металл, хорошо проводит электрический ток и тепло. Плотность 2,7 г/см3. Температура плавления равна 66О°С. Температура кипения 2450°С. Природный алюминий состоит из одного изотопа 2713А1.
Химические свойства.Алюминий — металл. На внешнем электронном слое у атома алюминия три электрона в состоянии ...3s23р1.В реакциях алюминий отдает эти электроны и превращается в положительно заряженный ион Al3+. Алюминий — сильный восстановитель, он находится в левой части электрохимического ряда напряжений металлов. Алюминий реагирует со многими простыми и сложными веществами.
Взаимодействие алюминия с простыми веществами.
1. Алюминий легко соединяется с кислородом при комнатной температуре, при этом на поверхности алюминия образуется оксидная пленка (слой А12О3). Эта пленка очень тонкая (~10-5 мм), но прочная. Она защищает алюминий от дальнейшего окисления, поэтому называется защитной пленкой: 4Al + 3О2 = 2А12О3Аналогичным уравнением выражается процесс сгорания порошка алюминия в пламени горелки.
2. При взаимодействии с галогенами образуются галогениды: 2А1 + 3С12 = 2А1С13 2А1 + 3Вr2 = 2А1Вr3
3. При взаимодействии с серой образуется сульфид: 2А1 + 3S = A12S3
4. При взаимодействии с азотом при высокой температуре образуется нитрид: 2А1 + N2 = 2A1N
5. При очень высокой температуре алюминий взаимодействует с углеродом и образует карбид: 4А1 + ЗС = А14С3
Взаимодействие алюминия со сложными веществами 1. Если с поверхности алюминия удалить оксидную пленку, то он активно взаимодействует с водой: 2А1 + 6Н2О = 2А1(ОН)3 + 3Н2↑
2. При высокой температуре алюминий реагирует с оксидами металлов, при этом образуется свободный металл и оксид алюминия. Взаимодействие алюминия при высокой температуре с оксидами металлов называется алюминотермией. Алюминотермию используют в металлургии для получения металлов: 2А1 + Fe2О3 = 2Fe + А12О3
3. Взаимодействие с разбавленными кислотами (НCl, Н2SО4):2Al + 6НС1 = 2А1С13 + 3Н2↑ 2AI + 3H2SО4 = A12(SО4)3 + 3Н2↑
4. Взаимодействие с концентрированной серной кислотой при нагревании: 8Al + 15H2SО4 = 4Al2(SО4)3 + 3H2S↑ + 12Н2О На холоду алюминий не взаимодействует с концентрированной H2SО4, так как пассивируется ею.
5. С концентрированной азотной кислотой алюминий не реагирует. Она пассивирует алюминий. Поэтому концентрированную азотную кислоту хранят в алюминиевых емкостях. С разбавленной азотной кислотой алюминий реагирует с образованием оксида азота (II): Al + 4HNО3 = AI(NО3)3 + NO↑ + 2Н2О
6. Взаимодействие алюминия со щелочами. Алюминий, как и другие металлы, образующие амфотерные оксиды и гидроксиды, взаимодействует с растворами щелочей. Рассмотрим механизм этого взаимодействия. Алюминий при обычных условиях, как уже было отмечено, покрыт защитной пленкой оксида А12О3. При погружении алюминия в раствор щелочи эта пленка растворяется: А12О3 + 2NaOH = 2NaAlО2 + Н2О, Освобожденный от защитной пленки алюминий, будучи активным металлом (Е° = —1,7 В), взаимодействует с водой подобно щелочным и щелочноземельным металлам: 2А1 + 6Н2О = 2А1(ОН)3 + 3Н2↑(I) Образовавшийся гидроксид алюминия, являясь амфотерным гидроксидом, взаимодействует со щелочью:А1(ОН)3 + NaOH = NaA1О2 + 2Н2О (2)
| Al | + NaOH | Na[Al(OH)4] + H2 |
| + HCl, H2SO4(ср. конц.) | Al3+ + H2↑ | |
| + H2SO4(конц.) | Al3+ + (SO2, S, H2S) | |
| + HNO3(ср. конц.) | Al3+ + NH4NO3 |
Складывая уравнения (1) и (2), получаем суммарное уравнение взаимодействия алюминия с раствором щелочи: 2А1 + 2NaOH + 2Н2О = 2NaA1О2 + 3H2↑
Кислоты:
Сильно разбавленная, а также концентрированные H2SO4 и HNO3 на Al не действуют, образуется защитная пленка толщиной 10-30 мкм.
С уксусной и фосфорной кислотами – не взаимодействуют.
Чистый алюминий довольно устойчив по отношению к HCl.
Кислородные соединения: А12О3, Al(OH)3 – амфотерны.
Al3+ + 3OH- ® Al(OH)3 ⇄ H3AlO3 ⇄ HAlO2 + H2O ⇄ H+ + AlO2-
1) Al(OH)3 + 3HCl = AlCl3 +3H2O
2) Al(OH)3 + NaOH = Na[Al(OH)4]
Соли Al подвергаются гидролизу, например, Al2S3, Al2(CO3)3 в воде не существуют, т.к. происходит полный гидролиз: 2AlCl3 + 3Na2CO3 + 6H2O = 2Al(OH)3↓ + 3H2CO3 ® (H2O + CO2↑)
Применение
1) Сварка: 8Al + 3 Fe3O4 = 4Al2O3 + 9Fe
2) Получение тугоплавких металлов(Cr, V, Mn): Cr2O3+2Al=Al2O3+2Cr
3) Искусственный рубин(Лазеры): Al2O3+Cr
4) Аммонал (взрывчатое вещество) – тончайший порошок Al в смеси с окислителями (NH4NO3).
5) Проводящий пленочный материал: (Пленки- вакуумные напыление )
ВЧ магнетронное распыление используют в качестве: проводников, контактных площадок, обкладок конденсатора
6) AlN-для изготовления преобразователей объемных и поверхностных акустических воль.(сапфир AlN, AlN-SiO2-SiO)
7) От предметов быта до ракетостроения.
Травление пленок алюминия
а) в щелочной среде (скорость травления 1 мкм в мин.)
Al+3K3[Fe(CN)6]+4KOH=3K4[Fe(CN)6]+K[Al(OH)4]
Al0+4OH- - 3ē = [Al (OH)4]-
3[Fe (CN)6]3- +3ē = 3[Fe(CN)6]4-
б) кислотные травители, растворы на основе HГалоген (HF, HCl), H2SO4, H3PO4, (HNO3 – пассивирует Al)
· t0 =60-700 C
Свойства
Все элементы подгруппы являются относительно химически инертными металлами. Характерны также высокие значения плотности, температур плавления и кипения, высокая тепло- и электропроводность.
Особенностью элементов подгруппы является наличие заполненного предвнешнего -подуровня, достигаемое за счёт перескока электрона с ns-подуровня. Причина такого явления заключается в высокой устойчивости полностью заполненного d-подуровня. Эта особенность обусловливает химическую инертность простых веществ, их химическую неактивность, поэтому золото и серебро называют благородными металлами.
Медь, серебро, золото. Изменение химической активности по группам d-элементов. Возможные валентности. Способы растворения металлов. Свойства соединений (способность к комплексообразованию, гидролиз солей).
Дата добавления: 2018-02-18; просмотров: 1010; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!