Синтезы кетонов из ацетоуксусного эфира
Лекция 7. Алкилирование енолятов в синтезе карбонильных соединений
Алкилирование енолятов в синтезе карбонильных соединений
Енолят-анионы образуются при депротонировании альдегидов и кетонов под действием оснований. Принципиально возможно два варианта генерирования енолятов, кардинально отличающихся условиями. Первый способ состоит в получении енолят-аниона в малой равновесной концентрации действием слабого основания при повышенных температурах. Второй вариант состоит в превращениикарбонильного соединения в 100 % енолят-анион под действием сильных ненуклеофильных оснований при низких температурах. Обработка енолятов алкилирующими агентами позволяет создавать новую С–С-связь, тем самым усложняя строение исходного карбонильного соединения. В идеализированном виде схема алкилирования енолятов приведена ниже:
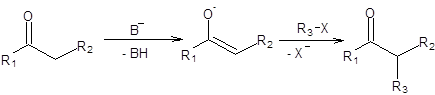
Схема 7.1
В общем случае алкилирование енолятов встречает ряд сложностей как на стадии генерирования енолят-аниона, так и на стадии его алкилирования:
1. Образование продуктов самоконденсации карбонильных соединений по реакции альдольной конденсации.
2. Образование смеси региоизомерных енолятов при енолизации несимметричных кетонов.
3. Образование смесей продуктов С-и О-алкилирования енолятов.
4. Возможность полиалкилирования енолятов.
5. Образование продуктов отщепления наряду с продуктами алкилирования енолятов.
|
|
|
Прогресс в проведении контролируемого алкилирования енолятов, достигнутый за несколько последних десятилетий, связан с применением сильных оснований, позволяющих генерировать 100% еноляты, избегая тем самым возможности побочного протекания альдольной реакции. Хотя многие классические методы, связанные с получением енолятов в малых равновесных концентрациях, до настоящего времени не утратили своего значения, они требует применения четко отработанных условий и далеко не универсальны.
Для кетонов енолизация под действием сильных оснований протекает быстрее, чем их самоконденсация по схеме альдольной реакции, что позволяет синтезировать 100% еноляты, тогда как для альдегидов скорость самоконденсации сопоставима со скоростью енолизации, что в общем случае не позволяет получать продукты алкилирования енолятов альдегидов с приемлемым выходом. Процесс енолизации альдегидов в общем виде выражается схемой, приведенной ниже:
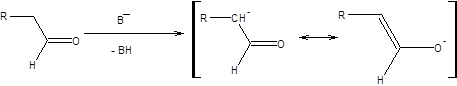
где В- – основание.
Схема 7.2
Процесс, протекающий параллельно самоконденсации альдегидов, обусловлен взаимодействием образующегося енолят-аниона (нуклеофил) с исходным альдегидом (электрофил) (схема 7.3).
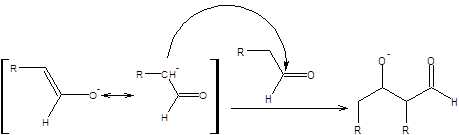
Схема 7.3
Кетоны также енолизуются в присутствии оснований, причем в общем случае возможно образование двух региоизомерных енолятов. Именно по этой причине возникает проблема региоселективного синтеза енолятов несимметричных кетонов:
|
|
|
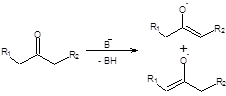
Схема 7.4
Енолизацию несимметричных кетонов можно проводить в кинетически и термодинамически контролируемых условиях. В термодинамически контролируемых условиях между двумя изомерными енолятами устанавливается равновесие, а в равновесной смеси преобладает термодинамически более стабильный енолят-анион. Так как с ростом числа алкильных заместителей термодинамическая стабильность двойной связи возрастает, то в условиях термодинамического контроля образуется более замещенный енолят - анион.
В условиях кинетического контроля равновесие между изомерными енолятами не устанавливается. В этих условиях соотношение изомерных енолятов определяется отношением скоростей их образования. Очевидно, что скорость депротонирования будет выше для наименее пространственно затрудненного a-углеродного атома, т.е. будет наблюдаться образование наименее замещенного енолята. Кинетическому контролю способствует пониженная температура и использование пространственно затрудненных сильных оснований, а также отсутствие избытка карбонильного соединения [1]. Для кинетически контролируемой енолизации необходимо использовать апротонные растворители, так как протонные растворители выступают как кислоты по отношению к сильным основаниям. Термодинамический контроль наблюдается при невыполнении хотя бы одного из условий, необходимых для осуществления кинетически контролируемой енолизации. Так использование слабых оснований для енолизации карбонильных соединений приводит к преимущественному образованию продуктов термодинамического контроля.
|
|
|
К числу пространственно затрудненных сильных оснований, необходимых для реализации кинетического контроля енолизации принадлежат амиды щелочных металлов различного строения. Примеры некоторых сильных ненуклеофильных оснований приведены на схеме 5 [2].
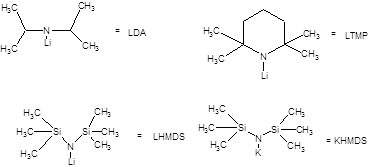
Схема 7.5
Примеры депротонирования кетонов в условиях кинетического контроля приведены на схеме 7.6 [3].
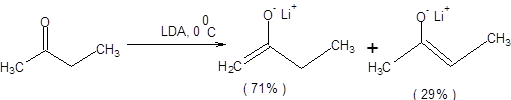
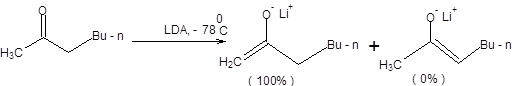
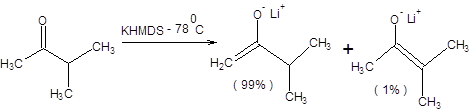
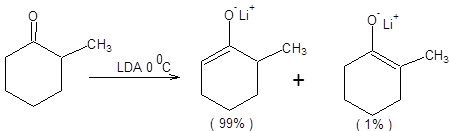
Схема 7.6
С ростом степени пространственной затрудненности оснований, применяемых для депротонирования кетонов, а также разницы в пространственных требованиях, предъявляемых алкильными группами кетонов, возрастает селективность кинетически контролируемой енолизации (схема 7.6).
|
|
|
Продукты термодинамически контролируемой енолизации образуются в избытке карбонильного соединения при длительном выдерживании реакционной системы даже в случае использования сильных ненуклеофильных оснований.
Енолят-анионы представляют собой типичные амбидентные нуклеофилы с двумя нуклеофильными центрами – атомами углерода и кислорода. Взаимодействие с электрофилами в общем случае может проходить как по атому углерода, так и по атому кислорода. Направление алкилирования и ацилирования енолятов сложным образом зависит от совместного действия следующих факторов:
1) Природы электрофильного реагента
2) Природы уходящей группы
3) Строения енолят-аниона
4) Природы противоиона енолят-аниона
5) Природы растворителя
Экспериментальные данные указывают, что последние два фактора не играют решающей роли в определении соотношения продуктов С-/O- замещения. В подавляющем большинстве случаев вопрос о направлении алкилирования и ацилирования енолят-анионов решается с помощью принципа ЖМКО. Согласно принципу ЖМКО, жесткие электрофилы преимущественно атакуют жесткий кислородный центр, а мягкие электрофилы – мягкий углеродный центр. К числу жестких электрофилов принадлежат ацилгалогениды, ангидриды карбоновых кислот, триалкилгалогенсиланы, a-хлорэфиры, a-хлорамины, сульфонилгалогениды, алкилсульфонаты. К мягким электрофилам относятся источники положительно заряженного галогена (N-бромсукцинимид, гипохлориты, галогены), сульфенгалогениды, селенгалогениды, алкилбромиды и алкилйодиды. Аллил- и бензилгалогенды и тозилаты проявляют свойства мягких электрофилов (схема 7.7).
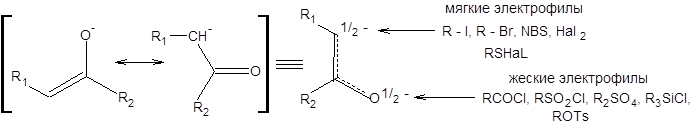
Схема 7.7
Примеры применения принципа ЖМКО с целью определения места атаки электрофила приведены на схеме 7.8.
А.Реакции с мягкими электрофилами
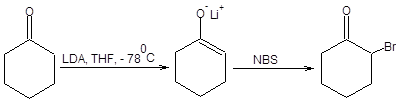

Б. Реакции с жесткими электрофилами
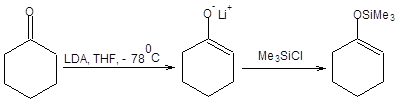
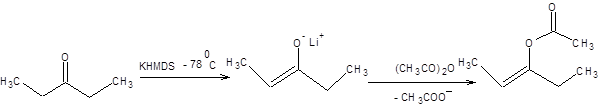
Схема 7.8
Наличие отработанных методов селективной енолизации кетонов и их алкилирования и ацилирования позволяет осуществлять тонкую «настройку» реакции в отношении селективного образования одного из возможных продуктов, как показано на схеме 7.9.
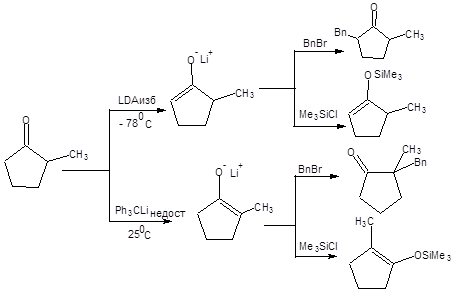
Схема 7.9
Несмотря на высокую предсказательную способность принципа ЖМКО при определении направления алкилирования и ацилирования енолятов существуют и другие факторы, не регламентированные этим принципом, которые необходимо учитывать. Например, следует учитывать стерические факторы при определении направления алкилирования. Электрофильная атака по нуклеофильному атому углерода чувствительна к стерическим факторам, тогда как нуклеофильный кислородный центр не предъявляет подобных пространственных требований. Увеличение объема нуклеофила приводит к снижению выхода продукта С-алкилирования, способствуя О-алкилированию. Так, неопентилхлорид алкилирует еноляты исключительно по атому кислорода.
Необходимо отметить, что алкилирующие реагенты аллильного и бензильного типа, занимающие важное место в синтетической химии, склонны образовывать продукты С-алкилирования. Так аллилбромид и аллилиодид алкилируют еноляты исключительно по атому углерода, а аллилтозилат – с преобладанием продукта С-алкилирования. Использование алкилйодидов и в меньшей степени алкилбромидов в качестве С-алкилирующих реагентов встречает сложности, связанные с возможностью диалкилирования. Для подавления диалкилирования рекомендуется использование алкилхлоридов вместо соответствующих алкилйодидов, а также применение малополярных, апротонных растворителей при возможно более низких температурах. Следует учитывать, что диалкилирование возможно лишь при использовании первичных алкилирующих агентов, уже применение вторичных галогенидов и тозилатов исключает диалкилирование ввиду существенных пространственных трудностей.
Диалкилсульфаты, алкилтозилаты и алкилтрифлаты являются в противоположность алкилбромидам и алкилиодидам жесткими алкилирующими агентами и способствуют образованию продуктов О-алкилирования с высокой селективностью. Алкилхлориды занимают промежуточное положение по своей жесткости между алкилбромидами и алкилтозилатами, и их использование в качестве алкилирующих агентов приводит к смеси продуктов С- и О- алкилирования (схема 7.10).
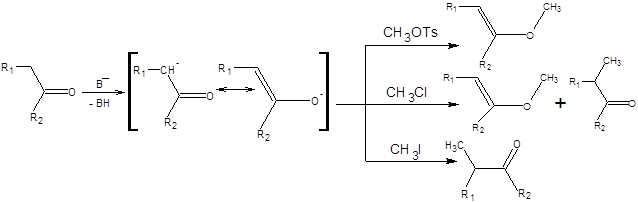
Схема 7.10
Алкилирование енолятов открывает удобный способ построения простой углерод-углеродной связи и может быть представлено в терминах ретросинтетического анализа схемой, приведенной ниже:
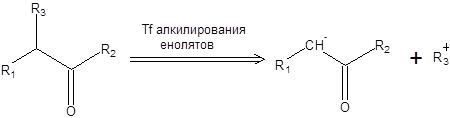
Схема 7.11
Сложные эфиры также депротонируются под действием сильных оснований. Еноляты сложных эфиров алкилируются и ацилируются исключительно по углеродному атому независимо от условий проведения реакции. Алкилирование енолятов сложных эфиров открывает широкие возможности для синтеза карбоновых кислот и их производных самого различного строения и рассмотрено в следующей главе.
В общем случае к образованию структур подобных енолят-анионам способны любые соединения, содержащие электроноакцепторные группы, резонансно стабилизирующие отрицательный заряд на α-углеродном атоме. Так возможно депротонирование амидов и нитрилов карбоновых кислот. В некоторых случаях вполне эффективно могут депротонироваться ангидриды кислот (например, в условиях реакции Перкина), однако хлорангидриды депротонируясь, затем элиминируют хлорид-ион превращаясь в кетены.
Распространение схемы «депротонирование с последующим алкилированием» на любые соединения, содержащие электроноакцепторные группы, привело к созданию группы методов позволяющих осуществлять расчленение С–С-связи в соответствии с общей схемой, приведенной ниже:
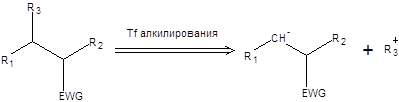
Схема 7.12
Самостоятельный интерес представляет алкилирование нитросоединений. Нитросоединения легко депротонируются под действием оснований, образуя при этом соли аци-формы. Соли аци-формы нитросоединений также подвергаются алкилированию и ацилированию. Алкилирование солей аци-формы нитросоединений происходит исключительно по атому кислорода с образованием нитроновых эфиров, которые термически неустойчивы и разлагаются выше 200С с образованием альдегидов и оксимов.
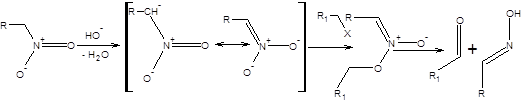
где Х – хорошая уходящая группа.
Схема 7.13
Алкилирование аци-формы нитросоединений особенно полезно для синтеза ароматических альдегидов из бензилгалогенидов, а также непредельных альдегидов из аллилгалогенидов (метод Хасса) (схема 7.14).
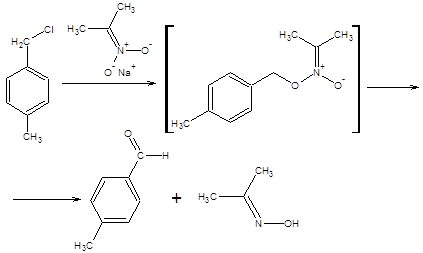
Схема 7.14
Совершенно иные синтетические возможности предоставляет двойное депротонирование нитросоединений, которое возможно осуществить действием на нитросоединение двукратного мольного избытка бутиллития. Образующиеся дианионы нитросоединений алкилируются и ацилируются исключительно по атому углерода. Последующее проведение реакции Нефа позволяет трансформировать полученное нитросоединение в кетон, как показано на схеме 7.15.
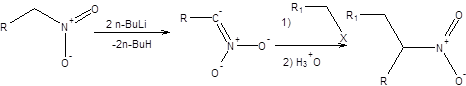
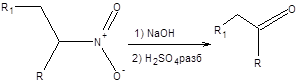
Схема 7.15
Как отмечалось выше, альдегиды не могут быть проалкилированны по обыкновенной схеме с приемлемым выходом ввиду чрезмерно высокой электрофильности их карбонильной функции. Для понижения электрофильности карбонильного атома углерода альдегидов их предварительно превращают в альдимины или диметилгидразоны [4]. Депротонирование азотистых аналогов альдегидов проводят при пониженной температуре, под действием LDA в качестве основания (схема 7.16).
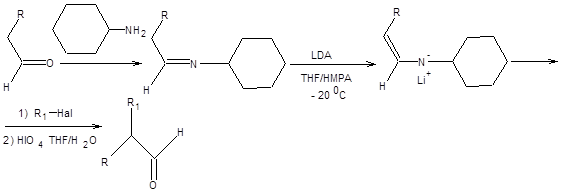
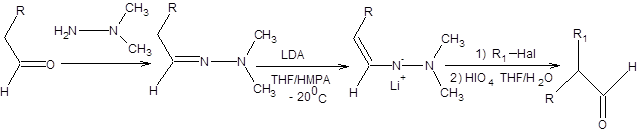
Схема 7.16
Алкилирование диметилгидразонов несимметричных кетонов происходит региоселективно по наименее замещенному α-углеродному атому. Примеры региоселективного алкилирования диметилгидразонов кетонов представлены на схеме 7.17.
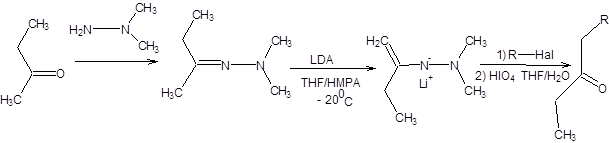
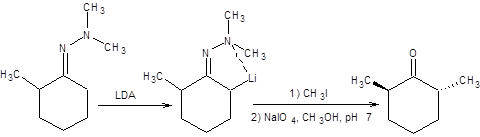
Схема 7.17
Все еноляты можно условно разделить на ионные и ковалентные в соответствии со степенью поляризации химической связи енолят-аниона с противоионом. Ионные и ковалентные еноляты обладают совершенно различной реакционной способностью. Так ионные еноляты – высокоосновные сильные нуклеофилы, а ковалентные, напротив, проявляют низкую основность при несколько меньшей нуклеофильности. Применение ионных енолятов ограничено в силу их несовместимости с электрофильными функциональными группами. Ковалентныееноляты, напротив, инертны к большинству функциональных групп в отсутствии катализа кислотами Льюиса. Некоторые аспекты реакционной способности ионных енолятов были рассмотрены выше.
К числу ковалентных енолятов относятся еноляты кремния и бора, получившие наибольшее значение в органическом синтезе. Ковалентные еноляты, равно как и ионные, могут быть получены в условиях термодинамического или кинетического контроля. Кинетические силиленоляты получают из кинетических ионных енолятов (схема 7.18).
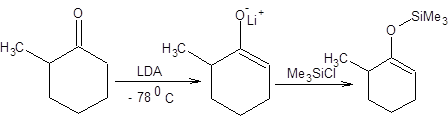
Схема 7.18
Термодинамический контроль синтеза силиленолятов предполагает использование слабых оснований и повышенных температур, а также значительную длительность процесса.
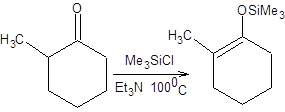
Схема 7.19
В отличие от нуклеофильных ионных енолятов, реагирующих с электрофилами по SN2 механизму, ковалентные еноляты существенно менее нуклеофильны и реагируют с электрофилами по АЕ механизму. Для проведения алкилирования силиленолятов электрофильную компоненту необходимо активировать, что достигается введением кислот Льюиса в реакционную систему. Механизм алкилирования силиленолятов представлен на схеме 7.20.
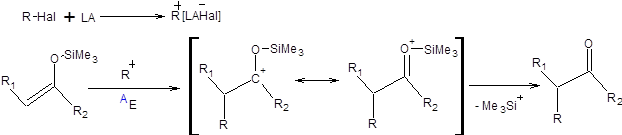
Схема 7.20
Переход от ионных енолятов к ковалентным енолятам сопровождается сменой типа катализа с основного на кислотный. Столь кардинальные изменения условий проведения реакции приводят к смене типа электрофильной компоненты в реакциях алкилирования. Для ионных енолятов, алкилирующихся по SN2 механизму необходимо использование первичных алкилгалогенидов и алкилсульфонатов, тогда как использование вторичных и особенно третичных алкилрующих агентов приводит к отщеплению. Для алкилирования ковалентных енолятов, напротив, предпочтительно использование вторичных и третичных галогенопроизводных и сульфонатов в присутствии кислот Льюиса (схема 7.21) [5].
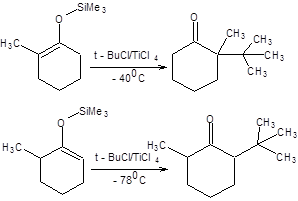
Схема 7.21
Ковалентные еноляты также возможно использовать как предшественники ионных енолятов, что позволяет смягчить условия получения последних. Для получения ионных енолятов из силиленолятов их обрабатывают эквивалентом фторид-иона, обладающего высоким сродством к кремнию. В качестве источника фторид-аниона в органических растворителях применяют тетрабутиламмонийфторид:
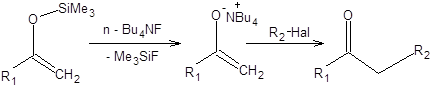
Схема 7.22
Синтезы кетонов из ацетоуксусного эфира
Ацетоуксусный эфир является достаточно сильной С–Н-кислотой и нацело депротонируется уже под действием этилата натрия. Повышенная С–Н-кислотность ацетоуксусного эфира связана с делокализацией отрицательного заряда между двумя электроноакцепторными заместителями. Для депротонирования ацетоуксусного эфира используются следующие основания: EtONa, t-BuOK, NaH, KH, LDA:
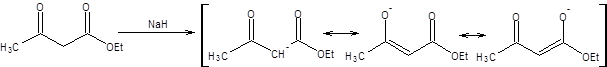
Схема 7.23
Алкилирование по атому углерода ацетоуксусного эфира с последующим кислотным гидролизом и декарбоксилированием приводят к образованию кетона. Моноалкилированный ацетоуксусный эфир может вторично подвергаться депротонированию с последующим алкилированием и кислотным гидролизом (схема 7.24).
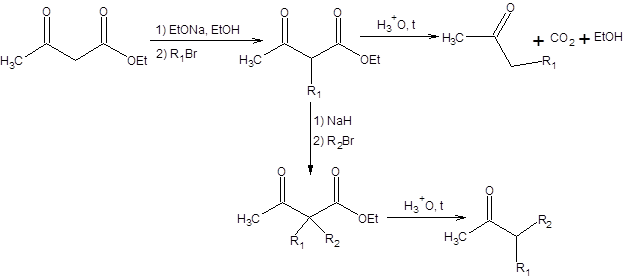
Схема 7.24
Другие возможности использования ацетоуксусного эфира в синтезе кетонов связаны с его способностью образовывать дианион при последовательном депротонировании сильными основаниями.
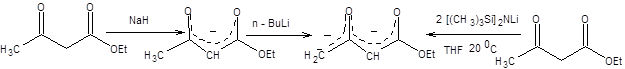
Схема 7.25
Дианион ацетоуксусного эфира в первую очередь алкилируется по первичному атому углерода как наиболее нуклеофильному, а затем по вторичному. Возможность селективного алкилирования дианиона ацетоуксусного эфира позволяет использовать его как общий предшественник кетонов в органическом синтезе (схема 7.26).
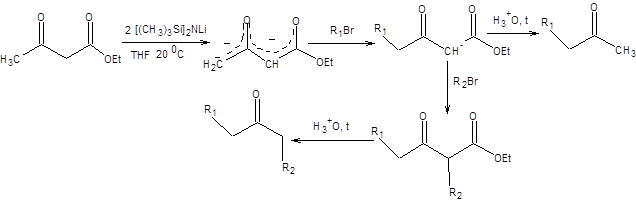
Схема 7.26
Дианион ацетоуксусного эфира можно с успехом применять и для построения циклов (схема 7.27).
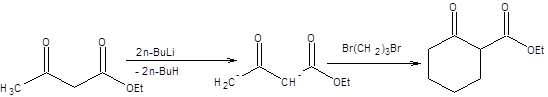
Схема 7.27
Применение ацетоуксусного эфира в качестве синтетического предшественника кетонов произвольного строения встречает трудности, связанные с невозможностью эффективно использовать вторичные и особенно третичные алкилирующие реагенты ввиду преобладания продуктов элиминирования. Указанные затруднения могут быть устранены, если алкилированию подвергать не енолят ацетоуксусного эфира, а его енол в условиях катализа кислотами Льюиса (схема 7.28).
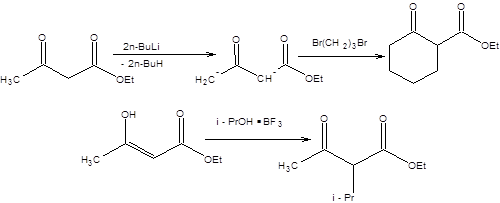
Схема 7.28
Ацетоуксусный эфир коммерчески доступен и образуется при самоконденсации этилацетата в присутствии этилата натрия.
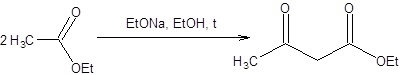
Схема 7.28
b-Кетоэфиры, являющиеся производными ацетоуксусного эфира, обладают комплексом схожих свойств. Общим методом синтеза b-кетоэфиров является С-карбоксилирование кетонов, действием диметилкарбоната магния (схема 7.29).
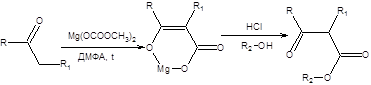 .
. 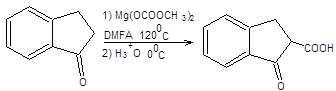
Схема 7.29
Аналогами β-кетоэфиров являются 1,3-кетосульфоксиды, обладающие значительной С–Н-кислотностью и получаемые перекрестной конденсацией сульфоксидов со сложными эфирами (схема 7.30).
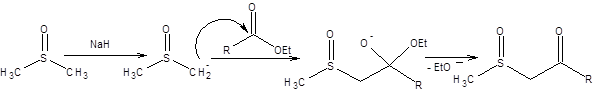
Схема 7.30
Высокая С-Н-кислотность 1,3-кетосульфоксидов позволяет депротонировать их сильными основаниями с образованием резонансно стабилизированного карбаниона, который далее подвергается алкилированию по традиционной схеме. 1,3-кетосульфоксиды восстанавливаются до кетонов амальгамой алюминия в воде или цинком в уксусной кислоте, что позволяет получать кетоны различного строения (схема 7.31).
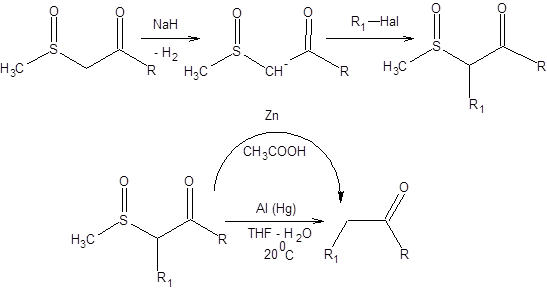
Схема 7.31
Дата добавления: 2021-05-18; просмотров: 261; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!