Кобальт-люминольная методика хемилюминесцентного определения пероксида водорода в реакционной смеси
Для выявления пероксида водорода в реакционной смеси при лакказном окислении фенолов применялась высокочувствительная методика, основанная на усилении хемилюминесценции кобальт-люминольной системы при введении H2O2 [46] (схема 2.1). Реакционная смесь, вводимая в кобальт-люминольную систему, разбавлялась в 105 раз, что обеспечивало отсутствие влияния на интенсивность хемилюминесценции примесей, также способных вызвать подобный эффект.

|
| Схема – Превращения люминола в присутствии ионов кобальта и пероксида водорода, приводящие к хемилюминесценции. |
Пероксид водорода в реакционной смеси при лакказном окислении гидрохинона
Концентрацию пероксида водорода в РС при лакказном окислении гидрохинона определяли хемилюминесцентным методом с использованием кобальт-люминольной методики (раздел 2). Реакцию лакказного окисления ГХ проводили в термостатируемом реакторе с барботацией ([ГХ]0 =1,36 мМ, [Е]0 = 20 мг/л, цитратный буфер рН 4,6, 35°С). Через 20 минут после начала реакции отобрали 0,05 мл реакционной смеси и развели дистиллированной водой в 10000 раз. В 5 мл кобальт-люминольной смеси ([люм] = 7,0 мкМ, [CoSO4] = 1,0 мкМ, карбонатный буфер 9.9) вводили по 5 мл разбавленной реакционной смеси или растворы пероксида водорода известной концентрации, что вызывает скачок на кривой ХЛ (рисунок 3.20). Сравнивая интенсивность этих пиков, можно сказать предварительно, что в данной реакционной смеси присутствует 1 мМ H2O2, то есть концентрация пероксида водорода соизмерима с начальной концентрацией ГХ.

|
| Рисунок - Влияние пробы реакционной смеси (стрелкой показан момент введения) на люминол-зависимую ХЛ. Карбонатный буфер рН 9.9; [люминол] = 7,0 мкМ, [CoSO4] = 1,0 мкМ, рН 9.9). |
По имеющимся данным о механизме реакции лакказного окисления органических субстратов в активном центре фермента, восстановление кислорода лакказой не сопровождается образованием пероксида водорода. В работе [4] подчеркивается, что в этом состоит преимущество лакказы для промышленного применения перед пероксидазами, для работы которых необходим пероксид водорода, в то же время приводящий к их дезактивации через несколько циклов. Однако не только восстановление кислорода, но и превращения феноксильного радикала, второго продукта лакказы, способны генерировать АФК, в том числе и H2O2, хотя бы по реакциям (3 – 4) [2].
 , ,
| (3) |
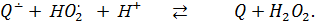
| (4) |
Из выводов:
Впервые обнаружен пероксид водорода в реакционной смеси в процессе окисления классического субстрата лакказ – гидрохинона – с помощью метода активированной кобальт-зависимой хемилюминесценции люминола. По предварительной количественной оценке содержание H2O2 в смеси соизмеримо с начальной концентрацией субстрата.
Синтез люминола_2
1. Синтез 3-нитрофталевой кислоты
H2SO4 85% (плотность 1.77) – 22,4 мл (0.4 моля), фталевый ангидрид (измельчен в ступке) – 5.2 г (0,035 моль), NaNO3 – 8 г (0,092 моль). Тщательно встряхнули, нагревали на водяной бане 1 час с обратным холодильником, периодически встряхивая. На ночь оставили.
1.1. Разделение 3 – и 4-нитрофталевой кислот
Н а следующий день добавлено 1-2 мл дист воды, монолит быстро разбился на взвесь, взвесь отфильтровали на фильтре Шотта. В фильтрате тоже выпал осадок, отфильтрован снова, обе фракции смешаны с небольшим количеством воды (3-5 мл), нагреты (полного растворения не происходит). Охладили во льду, выпало немного осадка, раствор мутный, отфильтрован через фильтр Шотта – фракция 1. Минут через 10 из раствора выпало много (на вид) осадка, отфильтрован - фракция 2. Через месяц из фильтрата отделена фракция 3 (предположительно 4-нитрофталевая кислота).
2. Получение 3-нитрофталгидразида (неудачная попытка)
2.55 г гидразинсульфата и 5.32 г ацетата натрия помещено в выпаривательную чашку, растворено в 5-10 мл горячей дист воды. Фракции 1 и 2 смешаны – 4.15 г 3-нитрофталевой кислоты. Добавлено в чашку. Жидкость становится ярко-желтой, сильный запах уксуса. Нагревали в песке с перемешиванием. Жидкость густеет и темнеет, выпадает р ыжий осадок. Вероятно, осадок плавится, так как потом это – темно-желтая густая масса, кипит, брызгает. Затвердевает, светлея. Масса очень твердая, измельчена стеклянной пробкой, оставлена для удаления летучих примесей в песке при ~ 160°С на 3 часа. Вероятно, была перегрета, цвет в конце – темно-желт ый (методики: не коричневый, а желтый или кремовый!). На будущее – греть при 150 или меньше.
После 3 часов масса промыта водой (нерастворима), сохнет.
Потом была сварена свежая 3-нитрофталевая кислота на серной кислоте из канистры, загрузка втрое больше. Продукт очищен тщательнее (его просто собирали меньше, не смешивая фракции). Попытка №2 проводилась именно с ним.
3. Получение 3-нитрофталгидразида попытка №2
1,85 г гидразинсульфата и 3,86 г ацетата натрия помещено в выпаривательную чашку, растворено в 5 мл горячей дист воды. 3 г 3-нитрофталевой кислоты добавлено в чашку. Жидкость становится ярко-желтой, сильный запах уксуса. Нагревали в песке с перемешиванием. Жидкость густеет и темнеет, выпадает осадок. Вероятно, осадок плавится, так как потом это – желтая густая масса. Затвердевает, светлея. Масса твердая, измельчена стеклянной палочкой, оставлена для удаления летучих примесей в песке при не более 160°С на 0,5 часа. Цвет в конце желтый, не коричневый.
4. Добавлено 5-10 мл дист воды, остается более светлый осадок и желтая жидкость. При фильтровании через ф. Шотта 100 частично потерян осадок. Собранный осадок бледно-кремового цвета высушен, выход 0,555 г.
5. После высыхания (жаркая погода) на воздухе фильтрата осталось довольно много неоднородного желтого осадка. Размешан с примерно 15 мл дист воды, отцентрифугирован. Осадок отделен, сохнет. Надосадочная жидкость испаряется. Жидкость потом вылита, осадок также утилизирован, потому что все получилось с первой фракцией и было не до того.
6. Первая фракция бледно-кремовый цвет 0,555 г восстановлена гидразинсульфатом. Согласно методике с сайта sam0delka(20 мл воды + 11 г гидразинсульфата (растворить) + 5 г 3-нитрофталгидразида, кипятить час) действовать не получилось, так как гидразинсульфат не растворялся в этом количестве воды. Растворимость (при сильном нагревании, до кипения) 1.22 г в 7-7.5 мл воды. Попутно гидразинсульфат перекристаллизован из воды (очень хорошо выпадает при охлаждении и выход перекристаллизации високий) и далее использовался очищенный.
Итак, восстановление 3-нитрофталгидразида.
1,22 г гидразина растворено при кипении в 7-7,5 мл дист воды, туда всыпан измельченный 3-нитрофталгидразид (0,555 г), который почти сразу и почти полностью растворяется, давая лимонно-желтый раствор. Раствор кипятится несколько больше часа (круглодонная колба, нагрев над печкой, обратный холодильник), в процессе выпадает бесформенная масса светло-желтого, песочного осадка. После охлаждения осадок отделяется на ф.Ш с бумажкой, фильтрат выливается. Осадок растворяем в 5% NaOH (1,38 мл 27% NaOH + 8,62 мл дист воды или 2 + 12,5 мл), ушло 7 мл раствора. Растворяется почти полностью, раствор темно-рыжий, похож на хромку. Фильтруем через ф.Ш без бумаги, отделяя якобы серу. Фильтрат нейтрализуем, прикапывая ледяную уксусную кислоту (ушло менее 1 мл) до нейтрального значения рН по индикаторной бумажке, сразу выпадает светло-желтый осадок. Осадок отделяется на ф.Ш, сушится на воздухе.
Продукт довольно хорошо растворяется в КБС рН 9.9 и дает свечение в смеси с кобальтом(2+), введение перекиси водорода приводит к скачку интенсивности.
7. Коричневое вещество (2,44 г) из п.2 (предположительно перегретый 3-нитрофталгидразид) также подвергнуто часовому кипячению с гидразинсульфатом (в соответствии с п.6). Полного растворения не происходило, осадок был, но имел другой вид. Жидкость была грязно-желтой. После кипячения РС разделена и продукт обработан аналогично п.6. Раствор в NaOH был темнее. Осадок темно-рыжего цвета оставлен сушиться на воздухе. Этот осадок позднее переосажден еще раз, остался примерно таким же на вид. Плохо сохнет (в стеклянном стакане, а не на бумаге).
8. Также проведено растворение в NaOH (прошло полностью, до ярко-желтого раствора) и дальнейшее осаждение уксусной кислотой «люминола», купленного в Синбиасе. Получен желто-коричневый осадок, оставленный сушиться на воздухе.
9. Нитрование фталевого ангидрида. Загрузка тройная – 15.6 г ангидрида, 24 г нитрата натрия, 67 мл серной кислоты из канистры. Нагревали на водяной бане 1 час с обратным холодильником, продукт сразу по охлаждении выпадать не начал, но на следующий день реакционная масса почти полностью затвердела. Туда добавлено порядка 20 мл дист воды, выпадает осадок густой, интенсивное выделение оксида азота. Полученная 3-нитрофталевая многократно промыта холодной дист водой (на будущее – промывать можно прямо в ф.Ш 16, не вытаскивая в стаканчик) ДОПИСАТЬ МЕТОДИКУ ОЧИСТКИ. Из фильтрата при его подвыпаривании в 2-3 раза выпала предположительно 4-нитрофталевая.
Тпл (капилляр, прикрученный резинкой к термометру, термометр над печкой) 3-нитрофтал = 200-210°С (220-222 лит), 4-нитрофтал – 160-165 (соответствует лит).
3-нитрофтал промыта еще раз на ф.Ш, оставлена сушиться на бумаге, с другой стороны пропечатанной (никогда так не делать!), на высохшем веществе остались чернила, оно промыто и еще один раз, высушено в бюксе. Выход 3-нитрофталевой 5.5 г, Тпл = 215°С.
10. Гидразинирование 5.5 г 3-нитрофталевой
3,49 г гидразинсульфата и 7,11 г ацетата натрия помещено в выпаривательную чашку, растворено в 10 мл горячей дист воды. 5,5 г 3-нитрофталевой кислоты добавлено в чашку. Жидкость становится ярко-желтой, сильный запах уксуса. Нагревали в песке с перемешиванием. Жидкость густеет и темнеет, выпадает осадок. Вероятно, осадок плавится, так как потом это – желтая густая масса. Затвердевает, светлея. Масса твердая, измельчена стеклянной палочкой, оставлена для удаления летучих примесей в песке при не более 160°С на 0,5 – 1 часа.
11. Восстановление 3-нитрофталгидразида – большая загрузка.
14,2 г гидразина растворено при кипении в 90-100 мл дист воды, туда всыпан измельченный 3-нитрофталгидразид (6,48 г), который почти не растворяется, давая лимонно-желтый раствор. Раствор кипятится несколько больше часа (круглодонная колба 200 мл, нагрев над печкой, обратный холодильник). После охлаждения осадок отделяется на ф.Ш с бумажкой, фильтрат выливается. Осадок растворяем в 5% NaOH (1,38 мл 27% NaOH + 8,62 мл дист воды или 2 + 12,5 мл), ушло 80 мл раствора. Растворяется почти полностью, раствор темно-рыжий, похож на хромку. Фильтруем через ф.Ш без бумаги, отделяя якобы серу. Фильтрат нейтрализуем, прикапывая ледяную уксусную кислоту (ушло менее 10 мл) до нейтрального значения рН по индикаторной бумажке, сразу выпадает светло-желтый осадок. Осадок отделяется на ф.Ш, сушится на воздухе. Сохнет плохо, несколько дней, пахнет укусом, темнеет. Вероятно, плохо отделился фильтрат и надо промыть водой еще раз.
12. Гидразинирование 4-нитрофталевой
1,44 г 4-нитрофталевой (предположительно – Тпл 160-165) + 0,888 г гидразинсульфата + 2.4 мл воды + 1,85 г ацетата натрия в фарфоровой чашке. Жидкость становится ярко-желтой, сильный запах уксуса. Нагревали в песке с перемешиванием. Жидкость густеет и темнеет, выпадает осадок. Вероятно, осадок плавится, так как потом это – желтая густая масса. Затвердевает, светлея. Масса твердая, измельчена стеклянной палочкой, оставлена для удаления летучих примесей в песке при не более 160°С на 0,5 – 1 часа. Все очень похоже на гидразнирование 3-нитрофталевой, может это тоже она, но грязнее (потому и Тпл меньше?). На следующий день маса измельчена (очень твердая), сколько соскреблось со стенок – выход продукта 2.4 г.
Дата добавления: 2019-02-22; просмотров: 292; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!