Дополнительные клинические проявления нейрофиброматоза I типа
- Когнитивные нарушения — затруднения освоения письма, чтения, математики. Часто сочетаются с умеренным снижением интеллекта (IQ<70)[24]. У больных часто отмечаются депрессия из-за стыда, вызываемого обезображиванием тела и лица нейрофибромами, боязнь общества;
- Эндокринные расстройства — феохромоцитома[41], нарушение роста и полового созревания[24];
- Эпилептические припадки[24];
- Снижение мышечного тонуса[24];
- Нарушения поведения[24];
- Заболевания, напрямую не связанные с вовлечением нервов, — стеноз почечной и легочной артерий, легочные кисты, интерстициальная пневмония, гипертрофия клитора, неправильное формирование отделов желудочно-кишечного тракта[1]. Стеноз почечной артерии, особенно в сочетании с феохромоцитомой, обуславливает развитие артериальной гипертензии
- Сирингомиелия[1]


МРТ левой голени: злокачественная опухоль оболочки большеберцового нерва при синдроме Реклингхаузена (NF-1).
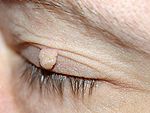
Нейрофиброма верхнего века.
Диагностика
Диагноз нейрофиброматоза I типа может быть поставлен при наличии у больного сочетания двух и более патогномоничных для заболевания симптомов[1][42][43]:
- шесть и более пятен цвета «кофе с молоком» диаметром свыше 5 мм у детей в препубертатном периоде и свыше 15 мм — в постпубертатном;
- наличие двух и более обычных нейрофибром, либо одной плексиформной нейрофибромы;
- гиперпигментация (по типу «веснушчатых гроздьев»[22]) подмышечной и/или паховой области;
- глиомы зрительных нервов;
- два и более узелка Лиша;
- костные аномалии (истончение кортикального слоя трубчатых костей, часто приводящего к формированию ложных суставов, дисплазии крыльев клиновидной кости);
- наличие нейрофиброматоза I типа у ближайших родственников.
Первичная диагностика заболевания производится как врачами общей практики, так и узкими специалистами (неврологами, дерматологами, офтальмологами, нейрохирургами, стоматологами и др.). Процесс развития клинической симптоматики при нейрофиброматозе I типа динамический[24].
Фенилкетонури́я (фенилпировиноградная олигофрения) — наследственное заболевание группы ферментопатий, связанное с нарушением метаболизма аминокислот, главным образом фенилаланина. Сопровождается накоплением фенилаланина и его токсических продуктов, что приводит к тяжёлому поражению ЦНС, проявляющемуся, в частности, в виде нарушения умственного развития.
В большинстве случаев (классическая форма) заболевание связано с резким снижением или полным отсутствием активности печёночного фермента фенилаланин-4-гидроксилазы, который в норме катализирует превращение фенилаланина в тирозин. До 1 % случаев фенилкетонурии представлено атипичными формами, связанными с мутациями в других генах, отвечающих за кодирование ферментов, обеспечивающих синтез кофактора фенилаланингидроксилазы — тетрагидробиоптерина (BH4). Заболевание наследуется по аутосомно-рецессивному типу.
Патогенез
Вследствие метаболического блока активируются побочные пути обмена фенилаланина, и в организме происходит накопление его токсичных производных — фенилпировиноградной и фениломолочной кислот, которые в норме практически не образуются. Кроме того, образуются также почти полностью отсутствующие в норме фенилэтиламин и ортофенилацетат, избыток которых вызывает нарушение метаболизма липидов в головном мозге. Предположительно, это и ведёт к прогрессирующему снижению интеллекта у таких больных вплоть до идиотии. Окончательно механизм развития нарушений функций мозга при фенилкетонурии остается неясным. Среди причин также предполагается дефицит нейромедиаторов мозга, вызванный относительным снижением количества тирозина и других «больших» аминокислот, конкурирующих с фенилаланином при переносе через гематоэнцефалический барьер, и прямое токсическое действие фенилаланина.
Диагностика
Производится полуколичественным тестом или количественным определением фенилаланина в крови. При нелеченных случаях возможно выявление продуктов распада фенилаланина (фенилкетонов) в моче (не ранее 10-12 дня жизни ребенка). Также возможно определение активности фермента фенилаланингидроксилазы в биоптате печени и поиск мутаций в гене фенилаланингидроксилазы. Для диагностики 2 и 3 типа, связанных с мутацией в гене, отвечающем за синтез кофактора, необходимы дополнительные диагностические исследования. В возрасте от 2-4 месяцев у больных появляются такие симптомы, как вялость, судороги, экзема, мышиный запах.
Лечение и профилактика
При своевременной диагностике патологических изменений можно полностью избежать, если с рождения и до полового созревания ограничить поступление в организм фенилаланина с пищей.
Позднее начало лечения хотя и даёт определённый эффект, но не устраняет развившихся ранее необратимых изменений ткани мозга.
Некоторые из современных газированных напитков, жевательных резинок и лекарственных препаратов содержат фенилаланин в форме дипептида (аспартам), о чём производители обязаны предупреждать на этикетке. Так, например, на этикетках ряда безалкогольных напитков компании «ПепсиКо» («Пепси», «Миринда» и др.), производимых в России, после указания состава и пищевой ценности 100 мл напитка приводится следующее предупреждение: «Содержит источник фенилаланина. Противопоказано применение при фенилкетонурии».
При рождении ребёнка в роддомах на 3-4 сутки берут анализ крови и проводят неонатальный скрининг для обнаружения врожденных заболеваний обмена веществ. На этом этапе возможно обнаружение фенилкетонурии, и, как следствие, возможно раннее начало лечения для предотвращения необратимых последствий.
Лечение проводится в виде строгой диеты от обнаружения заболевания как минимум до полового созревания, многие авторы придерживаются мнения о необходимости пожизненной диеты. Диета исключает мясные, рыбные, молочные продукты и другие продукты, содержащие животный и, частично, растительный белок. Дефицит белка восполняется аминокислотными смесями без фенилаланина. Кормление грудью детей, больных фенилкетонурией, возможно и может быть успешным при соблюдении некоторых ограничений[2][3].
Расчет диеты для больного ФКУ проводит врач с учетом потребности в фенилаланине и его допустимом количестве.
Допустимое количество фенилаланина для больных ФКУ[4]
| Возраст детей | Суточное количество фенилаланина (мк/кг массы тела) |
| До 2 мес. | 60 |
| 2-3 мес. | 60-55 |
| 3-6 мес. | 55-45 |
| 6-12 мес. | 45-35 |
| 1-1,5 года | 35-30 |
| 1,5-3 года | 30-25 |
| 3-6 лет | 25-15 |
| Старше 6 лет | 15-10 |
Некоторые (мягкие) формы заболевания поддаются лечению кофактором (тетрагидробиоптерином) пораженного фермента (фенилаланингидроксилазы). Разрабатываются новые подходы к лечению фенилкетонурии — использование заместительной терапии фенилаланинлиазой (PAL) — растительным ферментом, превращающим фенилаланин в безвредные метаболиты, и генотерапия на основе введения в организм вирусного вектора, содержащего ген фенилаланингидроксилазы. Эти методы пока не вышли из стен лабораторий. Атипичные формы не поддаются диетотерапии и лечатся только введением препаратов тетрагидробиоптерина или его синтетических аналогов (сапроптерин[5]).
Гомоцистинурия - (homocystinuria) - наследуемое заболевание, связанное с дефицитом фермента, вызванного избыточным содержанием гомоцистеина (промежуточного вещества, образующегося в ходе синтеза аминокислоты цистеина) в крови и наличием гомоцистина (окисленной формы цистеина) в моче. У больных отмечается задержка умственного развития, они имеют исключительно высокий рост, пальцы рук у них заметно удлиняются (вследствие усиленного роста костей); также у них наблюдается склонность к образованию кровяных сгустков в венах и артериях. В процессе лечения человек должен придерживаться строгой диеты
Причины гомоцистинурии
В результате снижения активности специального фермента печени (цистатионинсинтетазы) в организме ребенка накапливаются метионин и гомоцистин, оказывающие повреждающее действие на ряд систем организма (костную и центральную нервную системы), что приводит к развитию гомоцистинурии.
Проявления гомоцистинурии
Проявляются постепенно в течение первого года жизни ребенка в виде слабовыраженного отставания в весе и росте. При этом аппетит ребенка остается нормальным, функции желудочно-кишечного тракта не страдают, однако все попытки улучшить питание ребенка дополнительным введением белка в виде творога или кефира только усугубляют положение.
Отмечают позднее закрытие родничка, искривление конечностей, ребенок раздражителен, плаксив, у него нарушается сон, а дефицит массы тела нарастает.
Лечение гомоцистинурии
Существует две формы данного заболевания, одна из которых поддается лечению большими дозами (50-500 мг в сутки) витамина В6, а другая требует диетического питания. Диета должна быть малобелковой с низким содержанием метионина и дополнительным введением кальция, железа и витаминов.
 Количество метионина в лечебном питании составляет 29-45 мг на 1 кг массы тела ребенка, что достигается исключением (или снижением) из рациона продуктов животного происхождения.
Количество метионина в лечебном питании составляет 29-45 мг на 1 кг массы тела ребенка, что достигается исключением (или снижением) из рациона продуктов животного происхождения.
К специальным продуктам относятся так называемые желатиновые конфеты, содержащие смесь аминокислот, безбелковый хлеб, сахароза, декстрин-мальтоза.
Предлагаемые естественные продукты: желатин, молоко, кукурузные хлопья, рис, чечевица, арахисовое и кукурузное масла, картофель, другие овощи и фрукты.
Продукты с высоким содержанием метионина: творог, сыр, яйцо куриное, мясо говяжье, мясо кролика, мясо куриное, сельдь, треска, печень говяжья, почки говяжьи, соя, горох, мука пшеничная.
Продукты с низким содержанием метионина: молоко коровье, козье, женское, рис, морковь, свекла, картофель, горошек зеленый свежий и консервированный, помидоры, бананы, апельсины, мандарины.
Суточный набор продуктов (в граммах) для ребенка с гомоцистинурией (масса тела - 15 кг)
Критериями эффективности лечения являются нормальный уровень метионина в крови (0,01 г/л) и отсутствие гомоцистеина в моче.
Гала́ктоземи́я — наследственное заболевание, в основе которого лежит нарушение обмена веществ на пути преобразования галактозы в глюкозу (мутация структурного гена, ответственного за синтез фермента галактозо-1-фосфатуридилтрансферазы).
Галактоза, поступающая с пищей в составе молочного сахара — лактозы, подвергается превращению, но реакция превращения не завершается в связи с наследственным дефектом ключевого фермента.
Галактоза и её производная накапливаются в крови и тканях, оказывая токсическое действие на центральную нервную систему, печень и хрусталик глаза, что определяет клинические проявления болезни. Тип наследования галактоземии аутосомно-рецессивный.
Клиника
Заболевание проявляется в первые дни и недели жизни выраженной желтухой, увеличением печени, неврологической симптоматикой (судороги, нистагм (непроизвольное движение глазных яблок), гипотония мышц), рвотой; в дальнейшем обнаруживается отставание в физическом и нервно-психическом развитии, возникает катаракта. Тяжесть заболевания может значительно варьировать; иногда единственным проявлением галактоземии бывают лишь катаракта или непереносимость молока. Один из вариантов болезни — форма Дюарте — протекает бессимптомно, хотя отмечена склонность таких лиц к хроническим заболеваниям печени.
При лабораторном исследовании в крови определяется галактоза, содержание которой может достигать 0,8 г/л; специальными методами (хроматография) удается обнаружить галактозу в моче. Активность ферментов в эритроцитах резко снижена или не определяется, содержание ферментов увеличено в 10—20 раз по сравнению с нормой. При наличии желтухи нарастает содержание как прямого (диглюкуронида), так и непрямого (свободного) билирубина. Характерны и другие биохимические признаки поражения печени (гипопротеинемия, гипоальбуминемия, положительные пробы на нарушение коллоидоустойчивости белков). Значительно снижается сопротивляемость по отношению к инфекции. Возможно проявление и геморагического диатеза из-за уменьшения протеиносинтетической функции печени и уменьшения числа тромбоцитов — петехии.
Диагностика и дифдиагностика
Позитивные пробы на сахар и обнаружение галактозы в моче в первые дни жизни, а также уровень её в крови более 0,2 г/л требуют специального обследования ребёнка на галактоземию. Существуют специальные методы определения активности ферментов, превращающихся в галактозу, которые выполняются в централизованных биохимических лабораториях.
Дифференциальный диагноз проводится обычно с сахарным диабетом.
Тяжёлые формы заканчиваются летально в первые месяцы жизни, при затяжном течении на первый план могут выступать явления хронической недостаточности печени или поражения центральной нервной системы.
Лечение и профилактика
При подтверждении диагноза необходим перевод ребёнка на питание с исключением, главным образом, молока. Для этого разработаны специальные продукты: сояваль, нутрамиген, безлактозный энпит. Рекомендуются заменные переливания крови, дробные гемотрансфузии, вливания плазмы. Из лекарственных препаратов показано назначение оротата калия, АТФ, кокарбоксилазы, комплекс витаминов.
Показана высокая эффективность раннего выявления беременных в семьях высокого риска и внутриутробной профилактики, состоящей в исключении молока из диеты беременных.
Учёт семей риска позволяет рано, то есть ещё в доклинической стадии, подвергнуть специальному обследованию новорожденного и при положительных результатах перевести его на безлактозное вскармливание. Для раннего выявления предложены также специальные скрининг-программы массового обследования новорожденных.
С возрастом наблюдается ослабление этого специфического нарушения обмена.
СИНДРОМ УШЕРА
Синдром Ушера характеризуется врожденной нейросенсорной потерей слуха от умеренной до резко выраженной степени, вестибулярной гипофункцией и медленно прогрессирующим пигментным ретинитом. У некоторых больных могут наблюдаться умственная отсталость и поздние психозы.
Распространенность синдрома Ушера среди детей с глубокой глухотой составляет от 3 до 10 %. Вычислено, что один из 100 человек является носителем гена этого заболевания.
При классическом синдроме Ушера у всех больных имеются врожденные нейросенсорные нарушения слуха. У 90% из них степень потери слуха достигает 90 – 100 дб, у остальных – отмечается двусторонняя симметричная тугоухость II или III степени. Для всех больных характерны вестибулярные расстройства, выявляющиеся при проведении пробы на вращение. У многих при клиническом обследовании можно выявить нарушения равновесия или атактическую походку.
Изменение сетчатки начинаются постепенно. Первым симптомом болезни является нарушение темновой адаптации, которое проявляется в виде ночной слепоты. В дальнейшем пигментный ретинит медленно прогрессирует. Снижение остроты зрения становиться очевидным примерно к 10-летнему возрасту. К 40 –50 годам развивается полная слепота.
До настоящего времени не известны методы эффективного лечения синдрома Ушера. Поэтому, особенно большое значение приобретает профилактика этого тяжелого наследственного заболевания путем своевременного предупреждения семьи о риске повторного рождения больного ребенка. Это заболевание наследуется по аутосомно – рецесивному типу, то есть, считающие себя здоровыми родители больных детей являются скрытыми носителями гена болезни. Это может быть установлено или, хотя бы, заподозрено при помощи вестибулярных проб, электроретинографии и аудиометрии
Ранняя диагностика синдромаУшера чрезвычайно важна для правильного планирования коррекционно – педагогических мероприятий по обучению и воспитанию детей с данным заболеванием. Большую роль в этом играет проведение динамических углубленных офтальмологических обследований всех учащихся школ для детей с нарушением слуха, выявление среди них лиц с нарушением зрения, с пигментным ретинитом. Педагогам и родителям следует обращать внимание на детей с нарушением слуха, у которых ухудшается зрение.
Для определения правильного подхода к педагогической коррекции необходимо рано и точно диагностировать все имеющиеся у детей дефекты, определить характер и прогноз нарушенных функций, степень поражения зрения, слуха и состояния интеллекта.
Как правило, дети, имеющие синдром Ушера, начинают обучение в школах для глухих и слабослышащих. Снижение зрения у них диагностируется поздно. Из-за стойкой неуспеваемости они попадают в группу отстающих учеников. Между тем своевременное обучение по брайлеровской системе, развитие навыков осязания могли бы стать основой формирования словесной речи в письменной форме, что смягчило бы стрессовую ситуацию, связанную у глухого человека с потерей зрения.
Слухопротезирование лиц с синдромом Ушера проводится в зависимости от тяжести и характера поражения у них слуховой функции.
Перейти к: навигация, поиск
| Синдром Мартина — Белл | ||
| | ||
| Расположени FMR1 гена | ||
| МКБ-10 | Q99.2 | |
| МКБ-9 | 759.83 | |
| OMIM | 309550 | |
| DiseasesDB | 4973 | |
| eMedicine | ped/800 | |
| MeSH | D005600 | |

Синдро́м Ма́ртина — Белл (синдром ломкой X-хромосомы, fragile X mental retardation syndrome, FraX (от англ. fragile — хрупкий, ломкий)) — наследственное заболевание.
Развитие синдрома связано с экспансией единичных тринуклеотидов (ЦГГ) в Х хромосоме и приводит к недостаточной экспрессии белка FMR1, который необходим для нормального развития нервной системы. Существует четыре основных состояния хромосомного участка подверженного нарушениям при синдроме ломкой Х хромосомы, которые относятся к удлинению повторяющихся последовательностей CGG. Нормальное количество повторов (отсутствие синдрома) от 29 до 31. Премутация от 55 до 200 повторов (синдром не развивается). Полная мутация более 200 повторов (обычно от 230 до 4000) — проявляться синдром, промежуточное состояние или аллели серой зоны от 40 до 60 повторов[1].
Этиология
Синдром ломкой Х хромосомы развивается в результате мутации гена FMR1 в Х хромосоме. Мутация в этом гене встречается приблизительно у одного из 2000 мужчин и у одной из 259 женщин. Распространённость непосредственно заболевания приблизительно 1 из 4000 мужчин и 6000 женщин[2].
Экспансия повторяющихся CGG кодонов в такой степени приводит к метилированию части ДНК и как следствие фактическому прекращению экспрессии белка FMR1.
Подобное метилирование локуса FMR1 в хромосоме Хq27.3, как предполагают, является причиной сужения Х хромосомы, которая под микроскопом кажется хрупкой; в связи с этим синдром получил своё название.
Мутация гена FMR1 приводит к подавлению транскрипции белка FMRP. У здоровых индивидов FMRP, считают, регулирует значительную популяцию мРНК: FMRP играет важную роль в обучении и запоминании, а также принимает участие в развитии аксонов, формировании синапсов, появлении и развитии нервных связей[3].
Наследование
Синдром ломкой Х хромосомы сцепленное с полом доминантное заболевание с редуцированной пенетрантностью[4].
Мужчины имеют одну Х хромосому, соответственно если она содержит мутантный аллель у носителя развивается заболевание. Женщины несут две Х хромосомы, таким образом их шанс получить нормальный аллель удваивается. Женщина с мутантным геном FMR1 может иметь симптомы болезни ил быть здоровой. Несмотря на то, что вторая Х хромосома может служить резервной копией, только одна Х хромосома активна в каждой отдельной клетке, вследствие Х инактивации.
Мужчина с ломкой Х хромосомой не может передать её ни одному из сыновей, только всем дочерям. Женщина с одной мутантной хромосомой имеет одинаковые шансы передать ее как дочерям так и сыновьям с 50% вероятностью. Наследование синдрома ломкой Х хромосомы обычно увеличивается с каждым новым поколением, это явление получило название парадокса Шермана.
Патогенез
Это заболевание относится к болезням экспансии (экспансия — резкое увеличение числа копий повторяющихся участков молекулы ДНК (повторы) у индивидов в последующих поколениях родословной). Феномен экспансии числа тринуклеотидных повторов (ЦГГ) был впервые обнаружен как раз при молекулярно-генетическом исследовании этого синдрома.
Ранее диагноз синдрома Мартина-Белл основывался на данных клинико-генеалогического анализа и результатах цитогенетического исследования клеток больного, выращенных на специальной среде с дефицитом фолиевой кислоты. В случае обнаружения поломок X-хромосомы (ломкой X-хромосом, сайт Xq27.3) диагноз синдрома не вызывает сомнений.
В начале 90-х годов рядом исследователей было осуществлено секвенирование гена синдрома Мартина-Белл. Полученные результаты показали, что в основе клинических проявлений и цитогенетически выявляемой ломкости X-хромосомы при этом заболевании лежит многократное увеличение числа тринуклеотидных повторов ЦГГ. Оказалось, что у здоровых индивидов число этих повторов в X-хромосоме колеблется от 6 до 54, а увеличение этого числа свыше 200 повторов приводит к феномену ломкой X-хромосомы и клиническому проявлению заболевания. Предмутационное состояние - когда повторов ЦГГ от 55 до 200: заболевание у таких людей в типичной форме не проявляется, но высока вероятность того, что оно проявится у их потомков.
Экспансия тринуклеотидных повторов происходит во время гаметогенеза. Переход от состояния предмутации к полной мутации возможен только при передаче гена от матери, то есть «утяжеление» аллеля происходит во время овогенеза.
Клиническая картина
Мальчики рождаются с большой массой тела — от 3,5 до 4 кг. Первым признаком, который заставляет заподозрить заболевание — является макроорхизм при отсутствии эндокринной патологии. Также есть определённые фенотипические признаки: большая голова с высоким и широким лбом, длинное лицо с увеличенным подбородком, несколько уплощённая средняя часть лица, тупой, слегка клювовидно загнутый кончик носа. Уши большие, иногда оттопыренные, низко расположенные. Кисти и стопы широкие, дистальные фаланги пальцев также широкие, суставы имеют повышенную подвижность. Кожа нередко гиперэластична. Часто встречаются светлоокрашенные радужные оболочки, светлые волосы. Не обязательно встречаются все признаки — могут быть один или несколько.
Неврологическая симптоматика неспецифична, определяется как и у всех детей с умственной отсталостью. Наблюдается некоторая мышечная гипотония, дискоординация движений. Также могут быть глазодвигательные, пирамидные и экстрапирамидные нарушения.
Главным симптомом синдрома является интеллектуальное недоразвитие и своеобразная речь. Такие больные говорят быстро, сбивчиво, имеются выраженные эхолалии и персеверации (бормочущая речь). Также могут быть нарушения поведения в виде агрессивности, двигательной расторможенности. В качестве одной из частых психопатологических особенностей отмечена шизофреноподобная симптоматика, включающая в себя подпрыгивания, похлопывания руками, повороты вокруг своей оси, встряхивание кистями, «манежный» бег, разнообразные гримасы, монотонное хныканье.
Кроме вышеописанного у таких детей могут быть признаки раннего детского аутизма.
Диагностика
Синдром хрупкой Х хромосомы диагностируется путём определения количества ЦГГ повторов и их статуса метилирования с помощью эндонуклеазной рестрикции и саузерн блоттинга.
Лечение
Лечения для симптома ломкой Х хромосомы не существует, однако есть надежда, что дальнейшие исследования причин заболевания предоставят новые возможности терапии. В настоящее время, симптомы можно облегчить с помощью когнитивно-поведенческой терапии, специфического обучения, медикаментов и, при необходимости, лечения физических аномалий. Лица, имеющие случаи синдрома ломкой Х хромосомы в семье, должны получить генетическое консультирование при планировании беременности[5].
Поскольку в эксперименте обнаружение ломкости удалось обнаружить в среде, бедной фолатами, было предложено лечить таких детей фолиевой кислотой.
Эффект от лечения у детей выражен больше, чем у взрослых: пропадает агрессия, повышается внимание, улучшается моторика и речь.
Также пробуют лечить таких больных психостимуляторами.
ДЮШЕНА СИНДРОМ (Duchenne), характеризуется расстройством координации движений мозжечкового характера, выражающимся главным образом в изменении походки: она становится шатающейся, неуверенной, нерешительной, неправильной и медленной; больной ходит с широко расставленными ногами (походка пьяного человека); стоять неподвижно б-ной не может, качается. К расстройству походки часто присоединяются другие мозжечковые симптомы в различных комбинациях: дрожание в нижних конечностях, в туловище, голове и иногда в верхних конечностях, головокружение; эти симптомы увеличивают покачивание б-ного при ходьбе и могут вызвать его падение. Наблюдаются также следующие симптомы: нистагм, рвота, скандированная речь, расстройство почерка, более редко—косоглазие, астения без настоящих параличей и без особого понижения силы в каждой группе мышц, исследуемых в отдельности. Такой синдром наблюдается при поражении ver-mis 'а мозжечка
Синдром Дюшена является наследственным заболеванием, связанным с наличием модифицированного гена в половой Х-хромосоме.
Каждая клетка нашего организма (кроме половых) имеет 46 хромосом. Каждая хромосома в свою очередь состоит из тысяч генов. В генах и хромосомах содержится дезоксирибо-нуклеиновая кислота (ДНК), которая отвечает за информацию, передающуюся из поколения в поколение.
Каждый ген состоит из цепочки протеинов. Протеины — белки — являются строительным материалом человеческого opганизма.
Синдром Дюшена возникает вследствие наличия модифицированного гена в Х-хромосоме, которая присутствует как в мужском, так и в женском организме. Этот ген отвечает за выработку протеина, образующего здоровую мышечную ткань.
Девочки могут получить по наследству модифицированный ген, но, как правило, синдром Дюшена у них не развивается, так как в женском организме присутствуют две Х-хромосомы, одна из которых, здоровая, компенсирует отклонения во второй.
Результаты проведенных исследований показали, что синдром Дюшена у мальчиков может быть как врожденным, так и приобретенным вследствие изменений, произошедших в гене уже после рождения.
РАЗВИТИЕ БОЛЕЗНИ
По мере того как заболевание прогрессирует, симптомы становятся все более выраженными, так как мышцы уже почти не способны обеспечивать нормальные движения.
Мышцы рук постепенно слабеют, и ребенок с трудом может поднимать и удерживать предметы. Мышцы конечностей становятся ригидными, суставы теряют подвижность. Очень часто деформируются локтевые, тазобедренные и коленные суставы. Мышцы, поддерживающие позвоночник, атрофируются, вследствие чего позвоночник искривляется. Ребенку становится все труднее ходить. У некоторых детей возникают проблемы с учебой. Обучение дается им с большим трудом, они не успевают по всем предметам.
КАК МОЖНО ПОМОЧЬ РЕБЕНКУ?
Синдром Дюшена не поддается лечению, поэтому все, что можно сделать для таких детей, это помочь им научиться жить с этой болезнью.
Очень большое значение имеет психотерапия, которая способна помочь ребенку оставаться активным, не позволяя мышцам атрофироваться. Необходимо заниматься лечебной
гимнастикой для сохранения подвижности суставов. Чтобы предотвратить образование контрактур, могут использоваться шины и корсеты.
Для тех детей, которые хотя с трудом, но ходят, занятия ходьбой очень полезны.
В большинстве случаев у детей с синдромом Дюшена наблюдаются сложности с обучением, поэтому им требуется особое внимание и помощь. Лучше всего, если такой ребенок
будет обучаться в специализированном учебном заведении для детей-инвалидов.
Дата добавления: 2019-02-12; просмотров: 371; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!