Причины генных болезней (на примере эзимопатий)
Наследственные генные болезни обусловлены генными мутациями, изменяющими генетический код синтеза белков. Генные мутации возникают, когда последовательность нуклеотидов в ДНК гена изменяется. Существуют два основных класса генных мутаций: замена пар нуклеотидов, когда одна или несколько нуклеотидных пар в ДНК заменяются другими; мутация со сдвигом рамки считывания, обусловленные вставкой или выпадением одного или нескольких нуклеотидов. Замены пар оснований в нуклеотидной последовательности структурного гена часто приводят к замене одной аминокислоты в полипептидной цепи, определяемой одним геном. Мутации со сдвигом рамки считывания сильно изменяют последовательность аминокислот в транслируемом белке.
Нарушение синтеза белка при мутации соответствующего гена приводит к количественному или качественному изменению белка в организме. Генные мутации у человека являются причинами многих форм наследственной патологии. Если изменяется белок–фермент, выполняющий каталитическую функцию, то нарушается сложная цепь превращения вещества в организме: ген → фермент → биохимическая реакция → признак.
В биологической литературе такого рода изменения принято называть биохимическими мутациями, в медицинской литературе их называют наследственными дефектами обмена веществ или наследственными энзимопатиями. Функциональная неполноценность ферментной системы ведет к резкому нарушению определенного биохимического процесса или биохимическому блоку. Метаболический блок можно определить по накоплению в организме вещества, которое образуется на стадии, предшествующей этому блоку (рис. 1).
|
|
|
Выпадение одного единственного метаболического звена приводит к серьезным вторичным расстройствам обмена веществ и к множественным патологическим изменениям в организме.

|
| 1. Метаболические сдвиги при мутационной блокаде превращения одного вещества (Б) в другое (В) |
Степень снижения активности фермента может быть разной как при различных энзимопатиях, так и при данной энзимопатии. Снижение активности фермента или его отсутствие может быть обусловлено разными мутациями, происходящими в разных кодонах гена.
Кроме того, снижение активности фермента может быть связано с мутационным дефектом одного из компонентов ферментной системы. Следовательно, одни и те же биохимические изменения могут быть вызваны аллельными мутациями или мутациями в нескольких неаллельных генах. Таким образом, одна и та же энзимопатия может иметь несколько генетических форм. Это явление получило название генетической гетерогенности.
|
|
|
Широкая генетическая гетерогенность энзимопатии в значительной мере определяет изменчивость их клинических проявлений. Однако только особенностями мутационного гена нельзя объяснить неодинаковое проявление болезни у разных больных. В значительной степени ген проявляется во взаимосвязи с другими генами, вне зависимости от передающихся в семье. Эти гены могут усилить или затормозить проявление основного гена. Они могут изменить феномен наследственной болезни. Основной ген, в свою очередь, влияет на проявление других генов, благодаря чему у больного могут выявляться дополнительные, несвойственные основному заболеванию симптомы.
Таким образом, эффект мутантного гена можно рассматривать, как многоступенчатый процесс, первой ступенью которого является первичный биохимический дефект, второй – вовлечение в процесс других ферментных систем и развитие сложных метаболических расстройств, третий – формирование клинического феномена болезни.
Моногенные болезни наследуются в соответствии с законами Менделя и различаются типом наследования (таблица )
Таблица 1
Генные болезни, соответствующие определенным типам наследования
| Тип наследования | Заболевание | Локализация мутантного гена | Критерии наследования | ||
| аутосомно–доминантный
| Синдром Ваарденбурга | 2q37 (атрофия кортиева органа, врожденная глухота) | · Проявление признака у гетерозиготных носителей гена. · При анализе родословной признак выявляется в каждом поколении. · Пенетрантность патологических проявлений почти всегда ниже 100%. · Различная выраженность клинических проявлений не только между разными семьями, но и внутри каждой семьи. · Клинические признаки могут появиться не сразу после рождения, а спустя много лет. · Здоровые члены семьи не могут иметь больных детей. | ||
| Синдром Марфана | 15q21 (порок развития соединительной ткани) | ||||
| Синдром Реклингхау-зена (нейрофибро-матоз) | 22q12 (супрессор опухолевого роста) | ||||
| аутосомно–рецессивный
| Фенил-кетонурия (ФКУ) | 12q22 (нет синтеза фенилаланин-гидроксилазы) | · Мутантный ген проявляется только у гомозигот по рецессивному гену. · Если родители гетерозиготны, то вероятность рождения больного ребенка составляет 25%. · При анализе родословной мутантный ген проявляется не в каждом поколении. · Вероятность проявления мутантного гена возрастает в родственных браках. · Частота проявления мутантного гена у лиц женского и мужского пола одинакова.
| ||
| Гомоцисти-нурия | 21q22 (нет синтеза цистатионин-синтетаза) | ||||
| Галактоземия | 9р13 (нет синтеза галактозо-1-фосфат-уридилтрансфе-разы | ||||
| Синдром Ушера | 14q | ||||
| сцепленный с полом (рецессивный, сцепленный с X-хромосомой) | Синдром Мартина – Белла (ломкой Х–хромосомы) | Хq27 (? порок развития соединительной ткани) | · Мутантный ген (рецессивный) проявляется преимущественно у лиц мужского пола. · Если отец болен, мать здорова (фенотип, генотип), то все дочери будут гетерозиготными носительницами. Половая Х–хромосома от отца передается только дочерям. · Если отец здоров, мать фенотипически здорова (т.е. она носительница мутантного гена), то вероятность рождения больных сыновей составит 50%. · Если мутантный ген, локализованный в Х–хромосоме, является доминантным, то он проявляется и у мужчин, и у женщин. Частота заболевания женщин в популяции в 2 раза больше. | ||
| Синдром Дюшена (псевдогипертрофическая мышечная дистрофия | Хр16 (мутация гена дистрофина, кодирующего структурный белок сарколеммы). |
Синдром Ваарденбурга — наследственное заболевание. Имеет следующие клинические признаки: телекант (латеральное смещение внутреннего угла глаза), гетерохромия радужки, седая прядь надо лбом и врождённая глухота.
Телекант в сочетании с широкой и приподнятой спинкой носа и сросшимися бровями создаёт весьма своеобразный облик поражённых — «греческий профиль» . Очень характерны сросшиеся брови. Радужки либо различно окрашены (один глаз голубой, другой — карий), либо имеется сектор иного цвета в одной из радужек. У больных очень редко можно выявить весь набор типичных признаков: каждый симптом имеет свою степень экспрессивности. С наибольшим постоянством проявляется телекант — у 99 % носителей гена, широкая спинка носа — 75 %, сросшиеся брови — у 45 %, гетерохромия радужки — у 25 %, седая прядь или ранняя седина — у 17 % наблюдавшихся носителей гена.
Кроме указанных признаков, у больных иногда есть участки гипер- и депигментации на коже, пигментные изменения глазного дна. Седая прядь бывает уже у новорожденного, но затем эти депигментированные волоски часто исчезают. Нос часто имеет не только приподнятую спинку, но и гипоплазию крыльев. Патология конечностей включает такие аномалии, как гипоплазия кистей и мышц, ограничение подвижности локтевых, лучезапястных и межфаланговых суставов, слияние отдельных костей запястья и плюсны. Снижение слуха при этом заболевании врождённое, воспринимающего типа, связанное с атрофией преддверно-улиткового органа (кортиев орган). Глухота вызвана нарушениями спирального (кортиева) органа с атрофическими изменениями в спинальном узле и слуховом нерве. Синдром Ваарденбурга встречается с частотой 1:4000. Среди детей с врождённой глухотой составляет 3 %. Синдром определяется аутосомно-доминантным геном с неполной пенетрантностью и варьирующейся экспрессивностью. Ген локализован на хромосоме 2q37. При лечении в некоторых случаях показана косметическая хирургия телеканта. Лечение глухоты неэффективно.
Синдром Ваардербурга обычно наследуется по аутосомно-доминантному типу. Это значит, что одной копии измененного гена достаточно, чтобы вызвать данную патологию. В большинстве случаев один из родителей также имеет это заболевание. В редких случаях заболевание обусловлено спонтанно возникшей мутацией.
При некоторых разновидностях синдрома Ваарденбурга отмечается аутосомно-рецессивный тип наследования.
Синдром Марфана.
· Синдром Марфана (Болезнь Марфана) — аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани.
· Вызван мутациями генов, кодирующих синтез гликопротеина фибриллина-1, и является плейотропным.
· Характеризуется различной пенетрантностью и экспрессивностью.
· Распространенность в популяции составляет порядка 1 на 5000.
Клиника.
В классических случаях лица с синдромом Марфана высоки (долихостеномелия), имеют удлиненные конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой клетчатки.
Характерные изменений в органах опорно-двигательного аппарата (удлинённые трубчатые кости скелета, гипермобильность суставоварахнодактилия, деформации позвоночника (сколиоз, лордоз, гиперкифоз), деформация передней стенки грудной клетки (вдавленная грудь, «куриная грудь»), плоская стопа, высокое готическое небо, недоразвитие вертлужной впадины, врожденные контрактуры локтей и пальцев, мышечная гипотония);
Наблюдается патология в органах зрения (у половины больных диагностируется подвывих хрусталика; у лиц с выраженной миопией повышен риск отслойки сетчатки);
Сердечно-сосудистой системы (ССС) (пролапс митрального клапана отмечается в 80% случаев; со временем створки клапанов утолщаются, становясь гистологически миксоматозными; начинается дилатация корня аорты и прогрессирует с возрастом (у женщин отмечается более медленное прогрессирование), в конечном итоге может приводить к расслаивающейся аневризме аорты) - триада Марфана.
Прогноз.
Без лечения продолжительность жизни лиц с синдромом Марфана часто ограничивается 30—40 годами, и смерть наступает вследствие расслаивающейся аневризмы аорты или застойной сердечной недостаточности. В странах с развитым здравоохранением больные успешно лечатся и доживают до преклонного возраста.
Лечение.
Лечение — преимущественно симптоматическое, направлено на облегчение тех или иных проявлений заболевания.
Больным необходимо проходить расширенное ежегодное медицинское обследование с обязательным участием офтальмолога, кардиолога и ортопеда.
Перейти к: навигация, поиск
| Нейрофиброматоз I типа | ||
| | ||
| «Кофейные пятна» на коже. | ||
| МКБ-10 | Q85.0 | |
| МКБ-9 | 237.71 | |
| OMIM | 162200 | |
| DiseasesDB | 8937 | |
| MedlinePlus | 000847 | |
| eMedicine | derm/287 neuro/248 oph/338 radio/474 | |
| MeSH | D009456 | |

Нейрофиброматоз I (первого) типа (нейрофиброматоз с феохромоцитомой, болезнь фон Реклингхаузена, синдром Реклингхаузена, NF-1) — самое распространённое наследственное заболевание, предрасполагающее к возникновению опухолей у человека. Описан во второй половине XIX века рядом исследователей, в том числе в 1882 году учеником Рудольфа Вирхова Фридрихом фон Реклингхаузеном. Устаревшие названия — болезнь Реклингхаузена, периферический нейрофиброматоз и др. Является аутосомно-доминантным, встречается с одинаковой частотой у мужчин и у женщин, у 1 из 3500 новорождённых[1][2][3]. Другие типы нейрофиброматозов (на первую половину 2011 года выделяют 7 типов[4][5], из которых наибольшее клиническое значение имеют первые два) характеризуются наличием как сходных проявлений с I типом, так и отличий.
В половине случаев заболевание является наследственным, в половине — результатом спонтанной мутации. Частота мутаций генов, поломка которых приводит к нейрофиброматозу I типа, является самой высокой из известных для генов человека[1].
Для заболевания характерно появление множественных пигментированных пятен цвета «кофе с молоком», доброкачественных новообразований — нейрофибром, опухолей центральной нервной системы, костных аномалий, изменений радужной оболочки глаза и целого ряда других симптомов.
Нейрофиброматоз I типа проявляется рядом патогномоничных симптомов. К ним относят наличие пигментных пятен на коже цвета «кофе с молоком», нейрофибром, большинство из которых располагаются поверхностно на коже, узелки Лиша — гамартомы радужной оболочки глаза[25].
Проявления нейрофиброматоза I типа часто начинаются со сколиоза (искривления позвоночника), затем возникают трудности в обучении, проблемы со зрением и эпилепсия.
Нейрофибромы чаще локализуются по ходу периферических нервов. Однако может поражаться спинной и головной мозг, находят нейрофибромы на веках, конъюнктиве, в средостении, брюшной полости. В зависимости от расположения нейрофибромы могут вызвать различную клиническую симптоматику: судороги, нарушение функции черепных нервов и сегментов спинного мозга, паралич глазных мышц, птоз, сдавление органов средостения.
Нейрофибромы
Основная статья: Нейрофиброма
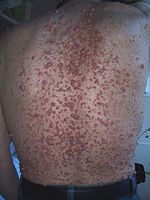

Множественные кожные нейрофибромы на спине больного нейрофиброматозом I типа
Для данного заболевания характерно появление большого количества нейрофибром, как кожных, так и плексиформных. Кожные нейрофибромы представлены небольшими доброкачественными и ограниченными новообразованиями. Они располагаются подкожно, растут на оболочках мелких нервов кожи. Плексиформные нейрофибромы развиваются на крупных нервах и приводят к нарушению их функций[26]. Также плексиформные нейрофибромы характеризуются своими большими размерами. Встречаются у 30 % больных нейрофиброматозом I типа[22].
Клинически повреждение нерва проявляется хроническими болями, онемением и/или параличами мышц.
Дата добавления: 2019-02-12; просмотров: 331; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!