Определение состава фосфолипидов в фосфолипидных продуктах и БАД
Метод основан на том, что пробу фосфолипидов, нанесенную на пластинку со слоем силикагель-гипс, разделяют в системе растворителей хлороформ-метанол-вода и идентифицируют полученные пятна.
Аппаратура и химическая посуда: весы лабораторные 2-го класса точности; шкаф сушильный, стеклянные пластинки размером 5х20, 10х20, 20х20 см со слоем силикагель-гипс; колба вместимостью 10 см3; микрошприц; хроматографическая камера; пульверизатор; эксикатор; цилиндр измерительный вместимостью 100 и 10 см3.
Реактивы, материалы: хлороформ; метанол; йод кристаллический; ацетон; нингидрин; висмут основной азотнокислый; кислота ледяная уксусная; йодид калия.
Техника выполнения. Приготовление пластинок с тонким слоем силикагеля, порядок хроматографирования осуществляют так же, как описано ранее.
Пробу обезжиренной смеси фосфолипидов, не содержащую нейтральные липиды, взятую на лабораторных весах, в количестве 0,05 г растворяют в 5 см3 хлороформа для получения примерно 1 %-ного раствора. На пластинку с тонким слоем сорбента микрошприцем наносят пробу полученного хлороформенного раствора фосфолипидов так, чтобы в пятне содержалось около 100 мкг фосфолипидов, что примерно соответствует 0,01 см3 1 %-ного раствора. Расстояние между точками с пробой-1,5 см.
Пластинку помещают в хроматографическую камеру, насыщенную парами системы растворителей хлороформ-метанол-вода в соотношении 65:25:4.
|
|
|
После окончания хроматографирования пластинку извлекают из камеры, быстро высушивают в вытяжном шкафу и приступают к обнаружению пятен.
Проявляют хроматограмму парами йода. Для этого пластинку помещают на 20...30 с в эксикатор, насыщенный парами йода. На хроматограмме появляются желтые или коричневые пятна, контуры которых необходимо тотчас обвести иглой.
Можно пластинки обработать соответствующими растворами для проявления пятен, пользуясь пульверизатором.
Индикатором на фосфолипиды, содержащие первичную аминную группу (фосфатидилэтаноламин, фосфатидилсерин), является 0,25 %-ный раствор нингидрина в ацетоне: после обработки появляются пятна пурпурного цвета.
Индикатором для обнаружения холинсодержащих фосфолипидов является реактив Драгендорфа, состоящий из смеси двух растворов: раствор I-1,7 г основного азотнокислого висмута, растворенный в 100 см3 20 %-ной уксусной кислоты; раствор II-40 г йодида калия, растворенный в 100 см3 воды. Перед использованием 20 см3 раствора I смешивают с 5 см3 раствора II и разбавляют 70 см3 воды.
Последовательность выхода групп фосфолипидов и соответствующие Rf для системы растворителей: хлороформ-метанол-вода (65:25:4) приведены в таблице 1.2.
|
|
|
Таблица 1.2 – Последовательность выхода групп фосфолипидов и соответствующие Rf
| Компоненты фосфолипидов | Величина Rf |
| Фронт-триацилглицерины, пигменты | - |
| Фосфатидные кислоты (ФК) | 0,74 |
| Лизофосфатидные кислоты (ЛФК) | 0,70 |
| Кардиолипин | 0,71 |
| Фосфатидилэтаноламин (ФЭА) | 0,62 |
| Фосфатидилглицерин (ФГЛ) | 0,48 |
| Фосфатидилхолин (ФХ) | 0,33 |
| Фосфатидилинозитол (ФИ) | 0,23 |
| Фосфатидилсерин (ФС) | 0,15 |
Контрольные вопросы
1. В чем сущность хроматографии.
2. На каких физико-химических свойствах основаны хроматографические методы анализа?
3. Как классифицируют хроматографические методы по каждому классификационному признаку?
4. Назовите характерные признаки хроматографии?
5. Область применения хроматографических методов?
6. В чем отличия различных методов хроматографии?
7. Назовите основные стадии тонкослойной хроматографии,
8. Что такое коэффициент распределения?
9. На чем основан количественный и качественный анализ при проведении тонкослойной хроматографии?
Лабораторная работа № 2
Определение жирнокислотного состава растительных масел методом газожидкостной хроматографии
Общие понятия. Газожидкостная распределительная хроматография является быстрым методом количественного анализа таких составляющих липидов, как углеводороды, алифатические спирты, жирные кислоты, стеролы и т.д.
|
|
|
Сущность метода заключается в том, что каждый компонент разделяемой смеси при определенной температуре в парообразном состоянии с определенной скоростью с помощью газа-носителя перемещается по колонке, заполненной порошкообразным носителем, пропитанным неподвижной фазой (нелетучая жидкость).
Хроматограмма смеси представляет собой кривую в виде ряда пиков, число которых соответствует числу разделяемых компонентов. Чем больше площадь пика, тем больше содержится данного вещества в пробе. Последовательность выхода компонентов зависит в основном от коэффициентов их распределения между жидкой неподвижной фазой и газообразной - подвижной. Поэтому каждый компонент характеризуется определенным временем удерживания от момента ввода пробы до появления максимума пика на хроматограмме. Время удерживания является непосредственно измеряемой величиной. Это функция физико-химических свойств компонента, длины колонки, температуры и скорости потока газа-носителя. Величиной, независящей от скорости потока газа-носителя, является удерживаемый объем, т.е. объем газа-носителя, который должен быть пропущен от момента ввода пробы до появления максимума пика компонента смеси на хроматограмме (рисунок 1).
|
|
|
|
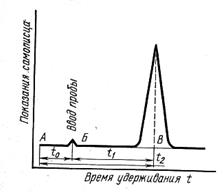 Рисунок 1- Хроматограмма
Рисунок 1- Хроматограмма
время - удерживаемым объемом для данного компонента. Этот показатель является специфической характеристикой каждого индивидуального вещества.
|
 Рисунок 2-Типовая схема газохроматографической установки:
1 - баллон с газом-носителем; 2 - редуктор; 3 - вентиль тонкой регулировки; 4 - осушительная трубка; 5 - манометр; 6 - подогреватель; 7 - детектор; 8 - самописец; 9 - узел ввода пробы; 10 - измеритель скорости потока
газа-носителя; 11 - термостат; 12 - хроматографическая колонка
Рисунок 2-Типовая схема газохроматографической установки:
1 - баллон с газом-носителем; 2 - редуктор; 3 - вентиль тонкой регулировки; 4 - осушительная трубка; 5 - манометр; 6 - подогреватель; 7 - детектор; 8 - самописец; 9 - узел ввода пробы; 10 - измеритель скорости потока
газа-носителя; 11 - термостат; 12 - хроматографическая колонка
Удерживаемый объем V рассчитывают по формуле (1)
V=ast2 , (1)
где аs-объемная скорость газа-носителя;
t2-время удерживания компонента.
Уточненное время удерживания определяется по уравнению (2) (рисунок 1)
t1=t2-t0 (2)
Тогда исправленный удерживаемый объем Vисп будет (3)
Vисп=ast2-ast0=V1-V0, (3)
где V0 - измеренный удерживаемый объем газа-носителя в период мертвого времени;
V1 - измеренный удерживаемый объем компонента.
Целесообразно относить удерживаемый объем вещества к удерживаемому объему стандарта Vст. Это отношение называется относительным удерживаемым объемом Vотн. Он зависит от природы и температуры жидкой фазы и мало зависит от изменения других параметров опыта (размера колонки, скорости потока и т.п.).
Относительный удерживаемый объем определяют по формуле (4) или (5)
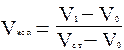 , (4)
, (4)
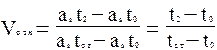 (5)
(5)
Таким образом, определяя время удерживания исследуемого компонента и время удерживания стандарта, можно рассчитать относительный удерживаемый объем.
Поскольку момент введения пробы автоматически отмечается на нулевой линии самописцем, то мертвое время можно не определять, тогда относительный удерживаемый объем при постоянном расходе газа-носителя будет зависеть только от отношения времени удерживания исследуемого компонента к времени удерживания стандарта, что определяется по формуле (6)
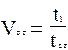 (6)
(6)
Любой аппарат для газовой хроматографии состоит из следующих главных узлов: источника газа-носителя с регулятором давления, устройства для введения образца, хроматографической колонки, термостата, хроматографического детектора и устройства для регистрации результатов анализа (рисунок 2).
Хроматографические колонки. В газовой хроматографии применяют колонки прямые, U-образные и спиральные.
Для их изготовления обычно используют трубки стеклянные, медные, полиэтиленовые и из нержавеющей стали. Внутренний диаметр колонки равен 2...6 мм, а длина - 5...20 м.
Носитель. Для хроматографии в системе газ-жидкость колонку наполняют измельченным инертным материалом-носителем, смоченным термостойкой жидкостью - неподвижной фазой. Носителем может быть любое твердое вещество, имеющее достаточно большую поверхность. В качестве носителя чаще всего применяют целит 545 (сорт кизельгура), измельченный огнеупорный кирпич С-22 (силоцел) и др.
Неподвижная фаза. Для успешного разделения смеси веществ важное значение имеет выбор неподвижной фазы. Она должна обеспечивать возможность многократного использования одной и той же колонки и полную воспроизводимость результатов.
При анализе смеси жирных кислот, спиртов и т.п. чаще всего применяют полярные жидкости-полиэфиры, например полиэтиленгликольсукцинат и др.
Газ-носитель (подвижная фаза). Газ, используемый в качестве подвижной фазы, должен быть инертным по отношению к твердому носителю и неподвижной фазе, к парам анализируемого образца и должен иметь низкую вязкость. Чаще всего в качестве газа-носителя применяют азот. Газ используют из баллонов, поток его поддерживается постоянным при помощи специального игольчатого вентиля. Скорость газа измеряют, определяя время вытекания заданного объема.
Детекторы. Для количественной оценки процесса газохроматографического разделения используют сигнал детектора, записываемый компенсационным ленточным самописцем.
Сигнал детектора - это результат измерения различий в теплопроводностях растворенных веществ в подвижной фазе (газе) или измерение электропроводности при ионизации растворенных веществ.
При исследовании жиров используют в основном дифференциальные детекторы, которые измеряют мгновенную концентрацию вещества в газе-носителе.
Термостат. Ввиду того что скорость продвижения компонентов смеси по хроматографической колонке зависит от температуры, колонку помещают в термостат (жидкостный, паровой, воздушный).
В настоящее время чаще всего используют воздушный термостат, позволяющий работать в области высоких температур.
Введение образца. Исследуемое вещество вводят с помощью инъекционных шприцев в верхнюю часть разделительной колонки.
Так как образец должен поступать в колонку в газообразном виде, то между дозатором и колонкой устанавливается подогреватель, обеспечивающий мгновенное испарение образца, температура в нем поддерживается на 30...500С выше температуры в колонке.
Идентификация. Ненасыщенные жирные кислоты на полярных фазах выходят после насыщенных кислот. Смеси жирных кислот с одинаковым числом углеродных атомов в молекулах выходят в следующем порядке: насыщенные-моноеновые--диеновые-триеновые.
Для идентификации жирных кислот пользуются данными относительных удерживаемых объемов жирных кислот (таблица 1), где в качестве стандарта принят удерживаемый объем пальмитиновой кислоты.
Для количественного определения рассчитывают площадь пика.
Для этого точно измеряют высоту пика и его ширину на половине высоты. Площадь пика рассчитывают, умножая высоту пика h на его полуширину b-hb (рисунок 3).
Таблица 3 - Величины относительных удерживаемых объемов
|
Кислоты |
Код кислоты | Относительный удерживаемый объем (неподвижная фаза-полиэтиленгликольсукцинат) при температуре, 0С | ||
| 200 | 203 | 220 | ||
| Каприловая | 8:0 | 0,096 | - | - |
| Капроновая | 10:0 | 0,181 | - | - |
| Лауриновая | 12:0 | 0,322 | 0,36 | - |
| Миристиновая | 14:0 | 0,565 | 0,60 | 0,588 |
| Пальмитиновая | 16:0 | 1,000 | 1,00 | 1,000 |
| Стеариновая | 18:0 | 1,750 | 1,66 | 1,620 |
| Олеиновая Элаидиновая | 18:1 цис 9 18:1 транс 9 | 2,000 1,960 | 1,93 - | - - |
| Линолевая | 18:2 цис 9,цис 12 | 2,450 | - | - |
| Линоленовая | 18:3 цис 9, цис 12, цис 15 | 3,160 | - | - |
| Арахидоновая | 20:4 цис 5, цис 8, цис 11, цис 14 | 5,440 | - | - |
| Бегеновая | 20:0 | 5,550 | 4,67 | 4,650 |
| Лигноцериновая | 24:0 | 9,890 | - | 8,890 |
|
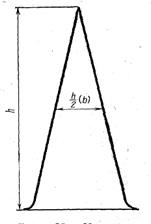 Рисунок 3 –
Измерение площади пика
Рисунок 3 –
Измерение площади пика
Смеси жирных кислот анализируют, переводя их перед разделением в метиловые эфиры, которые являются более летучими, чем свободные жирные кислоты и не обладают способностью димеризоваться. Кроме того, при хроматографировании эфиров облегчается разделение насыщенных жирных кислот от ненасыщенных с тем же числом углеродных атомов.
Эфиры насыщенных жирных кислот нормального строения выходят из колонки в порядке повышения их температуры кипения.
При постоянных определенных условиях работы время и порядок выхода метиловых эфиров жирных кислот из колонки всегда одинаков.
Непосредственному определению состава смеси жирных кислот ацилглицеринов методом газожидкостной хроматографии предшествует перевод жирных кислот в метиловые эфиры.
Дата добавления: 2018-11-24; просмотров: 632; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!