Осмотическое давление растворов ВМС.
Уникальные потребительские свойства полимеров, определившие стремительный рост и широчайшее распространение полимерных материалов, до 150 млн.т., с начала 20 века, обусловлены двумя фундаментальными физическими свойствами – высокоэластичностью и вязкоупругостью высокомолекулярных соединений. Высокоэластичность проявляется в больших обратимых деформациях – до 800% под действием малых нагрузок. Это свойство реализуется в таких полимерных материалах как каучук и резина. вязкоупругость подразумевает проявление полимерных свойств, присущих твердому телу и жидкости, т.е. сочетание обратимой и необратимой (течение) деформаций. Вязкоупругость приводит к пластичности и пониженной хрупкости полимерных материалов, называемых пластиками, поскольку необратимое перемещение макромолекул под нагрузкой вызывает релаксацию напряжения и предотвращает материал от разрушения. Чтобы правильно прогнозировать изменение свойств полимеров и изделий из них, а следовательно, и правильно направить их свойства при создании этих материалов и изделий, нужно знать закономерности изменения механических свойств от строения макромолекул. От природы цепи макромолекулы зависят такие важнейшие характеристики, как жесткость (гибкость) цепи, кристалличность, межмолекулярное взаимодействие. Эти свойства макромолекул определяют такие основополагающие для механических свойств полимера параметры, как деформация и прочность при разрыве.
|
|
|
Растворы ВМС
До середины 30-х годов ХIХ века существовали различные точки зрения на природу растворов ВМС. Одни исследователи считали, что растворы ВМС –истинные растворы, другие утверждали, что эти растворы являются типичными коллоидами, т. е. дисперсными системами. Разногласия объяснялись тем, что растворы ВМС обладают свойствами не только истинных растворов (самопроизвольность образования раствора, его термодинамическая устойчивость, молекулярная дисперсность, гомогенность), но и свойствами коллоидных растворов (неспособность молекул полимера проникать через полупроницаемую мембрану, низкое осмотическое давление, малые скорости диффузии молекул, светорассеяние). Когда же прояснился вопрос о размерах молекул ВМС, разногласия были исчерпаны. Оказалось, что свойства растворов ВМС, общие с коллоидными растворами, обусловлены соизмеримостью молекул полимеров и коллоидных частиц.
Полимеры, подобно низкомолекулярным веществам, в зависимости от условий получения раствора (природа полимера и растворителя, температура и др.) могут образовывать как коллоидные, так и истинные растворы. В связи с этим принято говорить о коллоидном или истинном состоянии вещества в растворе. Мы не будем касаться систем «полимер – растворитель» коллоидного типа. Рассмотрим только растворы полимеров молекулярного типа. Следует отметить, что вследствие больших размеров молекул и особенностей их строения, растворы ВМС обладают рядом специфических свойств:
|
|
|
1. Равновесные процессы в растворах ВМС устанавливаются медленно.
2. Процессу растворения ВМС, как правило, предшествует процесс набухания.
3. Растворы полимеров не подчиняются законам идеальных растворов, т. е. законам Рауля и Вант-Гоффа.
4. При течении растворов полимеров возникает анизотропия свойств (неодинаковые физические свойства раствора в разных направлениях) за счет ориентации молекул в направлении течения.
5. Высокая вязкость растворов ВМС.
6. Молекулы полимеров, благодаря большим размерам, проявляют склонность к ассоциации в растворах. Время жизни ассоциатов полимеров более длительное, чем ассоциатов низкомолекулярных веществ.
Термодинамика растворения ВМС. С термодинамической точки зрения растворение полимера, как любой самопроизвольный процесс, должен протекать с уменьшением свободной энергии системы (DG < 0).
|
|
|
Поскольку DG = DН – TDS, то уменьшению свободной энергии способствуют следующие два условия: DН < 0 (уменьшение энтальпийного фактора) и DS > 0 (увеличение энтропийного фактора). Растворение полярного полимера в полярном растворителе (неполярного – в неполярном) чаще всего сопровождается уменьшением внутренней энергии системы, так как растворение идет с выделением теплоты (DН < 0) вследствие гидратации (сольватации) макромолекул полимера.
Энтропия растворения высокомолекулярных веществ всегда во много раз выше энтропии растворения низкомолекулярных веществ. Это объясняется характерными особенностями химического строения макромолекул полимеров. Длинные гибкие макромолекулы могут принимать в растворе множество конформаций, которые мало различаются между собой по внутренней энергии. Известно, что состояние системы, которого можно добиться бóльшим числом микросостояний, обладает бóльшей термодинамической вероятностью W и, следовательно, характеризуется согласно уравнению S=klnW более высокой энтропией. Поскольку в растворе число возможных конформаций гибких макромолекул гораздо больше, чем в твердом полимере, то растворение полимера сопровождается значительным увеличением энтропии.
|
|
|
Энтропийный фактор особенно важен для неполярных полимеров с гибкими молекулами (каучук, поливинилацетат). Для таких полимеров увеличение энтропии обеспечивает соблюдение условия DG < 0 даже при увеличении энтальпийного фактора (DН > 0). В макромолекулах полярных ВМС, обычно обладающих жесткими цепями (поливиниловый спирт, белки), число возможных конформаций в растворе уменьшается, вследствие чего для этих полимеров возрастает значение энтальпийного фактора, т. е гидратация макромолекул.
Из выше сказанного следует, что образование растворов ВМС сопровождается уменьшением свободной энергии Гиббса. Следовательно, процесс растворения в данном случае идёт самопроизвольно и образующийся раствор будет термодинамически устойчив.
Набухание и растворение полимеров. Процесс растворения ВМС протекает самопроизвольно, но в течение длительного времени, и ему часто предшествует набухание полимера в растворителе. Полимеры, макромолекулы которых имеют симметричную форму, могут переходить в раствор, предварительно не набухая. Например, гемоглобин, печеночный крахмал – гликоген при растворении почти не набухают, а растворы этих веществ не обладают высокой вязкостью даже при сравнительно больших концентрациях. В то время, как вещества с сильно асимметрическими вытянутыми молекулами при растворении очень сильно набухают (желатин, целлюлоза, натуральный и синтетические каучуки).
Набухание – это увеличение массы и объема полимера за счет проникновения молекул растворителя в пространственную структуру ВМС. Причиной набухания является большая разница в размерах молекул растворяемого вещества и растворителя и, как следствие этого, большое различие в скоростях их диффузии. Поэтому при набухании вначале происходит практически односторонняя диффузия молекул растворителя в пространственную сетку полимера, имеющая ту же природу, что и осмос растворителя в осмотическую ячейку через поры полупроницаемой мембраны. Оба процесса вызываются стремлением системы к выравниванию концентраций компонентов.
Механизм набухания сводится к проникновению молекул растворителя в ближайшие слои полимера и сольватации соответствующих участков полимерной цепи. В результате этого макромолекулы «разрыхляются», что облегчает дальнейшее проникновение молекул растворителя и увеличение массы и объема полимера.
Различают два вида набухания: неограниченное, заканчивающееся полным растворением ВМС (например, набухание желатины в воде, каучука в бензоле, нитроцеллюлозы в ацетоне) и ограниченное, приводящее к образованию набухшего полимера – студня (например, набухание целлюлозы в воде, желатина в холодной воде, вулканизованного каучука в бензоле). Студень представляет собой пространственную сетку, состоящую из связанных между собой макромолекул полимера и заполненную молекулами растворителя.
Степень ограниченности процесса набухания и возможность самопроизвольного растворения определяются соотношением энергии связи в решетке полимера и энергии сольватации полимерной цепи с учетом энтропийного фактора.
Весь процесс набухания и растворения ВМС можно условно разделить на ряд стадий (рис. 43).
Рис 43. Последовательные стадии (а – г) растворения ВМС в низкомолекулярной жидкости
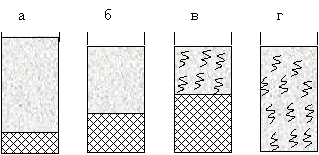
На начальной стадии (рис.43а) система состоит из двух компонентов: полимера и низкомолекулярной жидкости. Переход а®б характеризуется интенсивным проникновением молекул низкомолекулярной жидкости в структуру полимера и сольватацией полимерной цепи, сопровождающийся выделением теплоты (DН<0). Изменение энтропии по сравнению с энтальпийным фактором незначительно. При этом объем полимера возрастает, но общий объем системы полимер-растворитель уменьшается. Это явление называется контракцией, а выделение теплоты говорит о физико-химической природе процесса.
Переход б®в представляет собой начальный этап распределения макромолекул полимера по всему объему растворителя и характеризуется возрастанием энтропии системы вследствие роста числа возможных конформаций. Энтальпия системы если и изменяется, то незначительно. На данном этапе происходит обычно основное увеличение объема и массы полимера. Это результат дальнейшего проникновения молекул растворителя в полимерную сетку, ее разрыхление и связанное с этим частичное освобождение макромолекул. Отдельные макромолекулы начинают отрываться друг от друга и переходить в слой низкомолекулярной жидкости.
Ограниченное набухание заканчивается на стадии б или в образованием студня. Дальнейшее развитие процесса – неограниченное набухание – приводит к растворению полимера, т. е. образованию раствора ВМС (рис.43г). Переход в®г происходит в результате сил диффузии и характеризуется значительным увеличением энтропии системы. При этом макромолекулы ВМС равномерно распределяются по всему объему низкомолекулярного растворителя, образуя истинный раствор. Так как растворение полимеров главным образом обусловлено ростом энтропии, то и устойчивость растворов ВМС объясняется в основном энтропийным фактором.
Набухание и, следовательно, растворение ВМС зависят от природы растворителя и полимера, строения макромолекул полимера, температуры, присутствия электролитов, а также от рН среды (для полиэлектролитов).
Процессы набухания и растворения ВМС являются избирательными процессами. Другими словами для образования раствора ВМС необходимо его сродство с растворителем (лиофильность). Неполярные полимеры хорошо набухают (растворяются) в неполярных растворителях (каучук в бензоле или бензине) и не набухают в полярных. Полярные полимеры лучше набухают (растворяются) в полярных жидкостях (белок в воде) и не набухают в неполярных. Ввиду сродства полимера с растворителем, при набухании и растворении большая часть растворителя “связывается” в сольватные (гидратные) оболочки. Особенно это характерно для полярных макромолекул в водной среде. И поскольку макромолекулы обладают большой поверхностью, то для неограниченного набухания (растворения) даже в лиофильной системе требуется достаточное количество жидкости. Иначе процесс набухания может остановится на стадии ограниченного набухания, т. е. образования студня.
Существенную роль в набухании играет строение макромолекул полимера. Например, полимеры с длинными жесткими цепями и большим количеством полярных групп хорошо набухают, но не растворяются даже в соответствующем растворителе (целлюлоза в воде). Если полимер растворяется в жидкости не достаточно хорошо, то также образуется студень.
Температура на эти процессы влияет в соответствии с принципом Ле Шателье. Поскольку набухание сопровождается выделением теплоты на первом этапе, то с повышением температуры степень набухания, а так же растворимость полимера, уменьшаются. На второй стадии набухание может стать эндотермическим процессом. Следовательно, в этом случае набухание с возрастанием температуры увеличивается. Например, если в холодной воде желатина набухает ограниченно, то с повышением температуры – неограничено, т. е. растворяется. При охлаждения полученного раствора снова образуется студень. Однако скорость набухания (растворения) полимеров с увеличением температуры растет ввиду увеличения скорости диффузии.
Действие ионов электролитов на набухание полярного ВМС связано с их способностью к гидратации. Поскольку анионы гидратируются больше, чем катионы, то последние влияют на набухание этих полимеров незначительно. По способности уменьшать набухание анионы располагаются в так называемый лиотропный ряд, или ряд Гофмейстера (при одном и том же катионе):
CNS– < J– < Br– <  < Cl– < CH3COO– <
< Cl– < CH3COO– < 
Ионы CNS– усиливают набухание вследствие того, что слабо гидратируясь, они хорошо адсорбируются на макромолекулах ВМС. А ионы процесс набухания тормозят, так как сульфат – ионы сильнее всех анионов этого ряда гидратируются, уменьшая этим количество “свободной” (не связанной в гидратные оболочки) воды.
Влияние рН среды особенно значительно для высокомолекулярных электролитов (белков, нуклеиновых кислот, производных целлюлозы и крахмала). Минимум набухания отмечается в изоэлектрической точке, поскольку в ней суммарный электрический заряд макромолекул белков и, соответственно, степень их гидратации минимальны. При более низких или более высоких значениях рН увеличивается ионизация функциональных групп, что приводит к расталкиванию одноименно заряженных участков полимерной цепи и её разрыхлению. Вследствие этого молекулы воды легче проникают в пространство между цепями, что отражается на величине набухания в сторону ее увеличения.
Примером влияния рН на набухание является отек ткани человека, вызванный пчелиным или муравьиным ядом, имеющим кислую реакцию.
Количественной характеристикой ограниченного набухания полимеров является степень набухания a, определяемая отношением приращения массы (m – m0) или объема (V – V0) полимера к его первоначальной массе m0 (к объему V0):  или
или 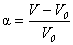 , (27)
, (27)
где m – масса (V – объем) набухшего полимера.
Набухание полимеров сопровождается возникновением давления, которое назвали давлением набухания (» 5×105–10×105 Па). Механизм его возникновения подобен механизму возникновения осмотического давления. Это давление легко обнаруживается, когда какое-либо препятствие мешает увеличению объема полимера.
Осмотическое давление растворов ВМС.
Осмотическое давление растворов низкомолекулярных и высокомолекулярных веществ определяется теоретически уравнением Вант-Гоффа:
Росм. = CRT, (29)
Осмотическое давление можно выразить и по другому:
Росм. , (29а)
где С – концентрация растворенного вещества в г/л, а М – молярная масса растворенного вещества.
Таким образом, уравнение (29а) можно использовать для определения молярных масс. Рассмотрим систему, в которой раствор, содержащий 20 г гемоглобина в 1 л, помещен в правый сосуд, а чистая вода - в левый, отделенный от правого полупроницаемой мембраной (рис.44). После достижения равновесия высота столба воды в правом сосуде на 7,78 см превышает высоту в левом сосуде.
Температура системы поддерживается постоянной, равной 2980 К. Какой же будет молярная масса гемоглобина?

Рис. 44. Схема прибора для демонстрации осмотического давления
Для ее определения сначала рассчитаем осмотическое давление раствора. Поскольку  , где А – площадь сечения трубки (м2); h – разность высот менисков (0,0778 м); r – плотность раствора (103 кг/м3); g – ускорение свободного падения (9,807 м/с2).
, где А – площадь сечения трубки (м2); h – разность высот менисков (0,0778 м); r – плотность раствора (103 кг/м3); g – ускорение свободного падения (9,807 м/с2).
Таким образом:
Росм.= 0,0778 м×103 кг/м3×9,807 м/с2 = 762,46 кг/м×с2 = 762,46 н/м2
Согласно уравнению (29а).

С повышением концентрации ВМС (кроме глобулярных полимеров) их осмотическое давление перестает подчиняться закону Вант-Гоффа и растет быстрее (рис.45). Причиной отклонений от закона Вант-Гоффа является относительная независимость теплового движения отдельных сегментов линейных макромолекул ВМС. Каждая макромолекула ведет себя как совокупность нескольких молекул меньшего размера. Это и проявляется в увеличении осмотического давления. Для расчета осмотического давления растворов ВМС Галлер предложил уравнение:
Росм.  , (30)
, (30)
где, С – концентрация раствора ВМС, г/л; М – молярная масса ВМС, г/моль; b – коэффициент, учитывающий гибкость и форму макромолекулы в растворе.
Коэффициент b зависит от природы растворителя и растворенного вещества, но не зависит от молярной массы растворенного полимера. С увеличением длины макромолекулы и разветвленности цепи величина b растет. Увеличение эффективного числа подвижных единиц (кинетически активных единиц) в растворе учитывается дополнительным слагаемым b С2. При небольших концентрациях полимера значение слагаемого невелико и уравнение Галлера переходит в уравнение Вант-Гоффа. Уравнение Галлера можно преобразовать в уравнение прямой, разделив обе части его на С:  (31)
(31)

Рис.45. Зависимость осмотического давления от концентрации раствора ВМС:
а – теоретическая кривая в соответствии с уравнением Вант-Гоффа;
б – экспериментальная кривая

Рис.46. График зависимости Росм /С от
концентрации С
Измерив осмотическое давление растворов с различной концентрацией С, можно построить графическую зависимость величины Росм./С от С и найти значение молярной массы М полимера и коэффициента b (рис.46).
Осмометрическим методом обычно пользуются для определения молярных масс ВМС в интервале от 10000 до 70000 г/моль. Нижний предел зависит от свойств мембран, а верхний определяется той чувствительностью, при которой можно измерять осмотическое давление. Погрешность результатов измерений осмотического давления растворов ВМС может быть связано с присутствием в растворе низкомолекулярных электролитов. Чтобы предотвратить влияние последних, раствор ВМС предварительно диализуют.
Следует заметить, что молярные массы ВМС нельзя определить традиционным криоскопическим методом. Это объясняется тем, что разбавленные растворы ВМС в общем случае не подчиняются закону Рауля. Поэтому, кроме описанного выше осмометрического метода разработаны и другие методы определения молярных масс ВМС: химический, вискозиметрический, методы седиментации и светорассеяния растворов, метод гель-фильтрации, электрофоретические и т. д. Ни один из перечисленных методов не является универсальным, так как каждый из них можно применять только при определенном диапазоне молярных масс полимеров.
Вязкость растворов ВМС
Вязкость жидкости можно определить как сопротивление жидкости передвижению одного её слоя относительно другого. Любое перемещение одной части жидкости относительно другой тормозится силами притяжения между её элементами. Иначе говоря, вязкость жидкости характеризует внутреннее трение, возникающее при перемещении слоев жидкости относительно друг друга.
Основы теории вязкости. При теоретическом рассмотрении вязкости жидкость представляется в виде бесструктурной непрерывной среды. Если приложить силу к жидкости, она начинает течь. Для жидкостей характерны два основных типа течения: ламинарное и турбулентное. Ламинарным называют течение жидкости в виде параллельных слоев, не перемешивающихся между собой. Такое течение существует до тех пор, пока величина градиента скорости не слишком велика. При увеличении градиента скорости слои жидкости образуют завихрения и перемешиваются. В таких случаях ламинарный поток переходит в турбулентный и ситуацию трудно трактовать как теоретически, так и экспериментально. Рассматриваемые нами закономерности вязкости будут относиться только к ламинарному режиму течения.
Рассмотрим два примыкающих объемных элемента какой-то жидкости. Если один из объемных элементов перемещается относительно другого под действием внешней силы, то между ними возникают силы, которые будут препятствовать такому перемещению, стараясь вернуть объемные элементы в их положение равновесия. Эта препятствующая сила называется силой (F) внутреннего трения (сопротивления).

Рис.47. Определение коэффициента вязкости h.
Сила сдвига между двумя элементами равна
h(dv/dx) × S, что определяет, таким образом, величину h
Чтобы определить вязкость количественно, можно воспользоваться рис.47. Предположим, что один из объемных элементов жидкости, представленных на этом рисунке, движется со скоростью dvотносительно второго элемента. Можно ожидать, что сила трения будет пропорциональна относительной скорости dv и площади контакта S между соседними элементами объема. Она будет обратно пропорциональна расстоянию dx между центрами этих элементов. Константа пропорциональности, связывающая силу трения и эти переменные, называется коэффициентом вязкости или просто вязкостью h. Обозначив силу трения через F, получим:
 (32)
(32)
Это определение вязкости первоначально дал Ньютон. Оно является микроскопическим, выраженным через величины, которые нельзя измерить.
Единицей вязкости служит ньютон-секунда на квадратный метр (Н×с/м2) или паскаль-секунда (Па×с); раньше за единицу вязкости принимали пуаз:
1 пуаз=0,1 Па×с.
Особенности вязкости растворов полимеров. Коэффициент вязкости, вычисленный по уравнению (32), определяется как константа пропорциональности и, таким образом, не зависит ни от приложенного давления, ни от градиента скорости (в условиях равномерного ламинарного течения). Жидкость, подчиняющаяся закону Ньютона, называется ньютоновской. Растворы ВМС не являются таковыми, поскольку величина их вязкости (h) зависит от градиента скорости. Дело в том, что для растворов ВМС само явление течения обусловливает ориентацию растворенных макромолекул (рис. 48).

Рис.48. Изменение структуры растворов ВМС при увеличении градиента скорости
При увеличении градиента скорости макромолекулы ориентируются вдоль оси потока, в связи с чем вязкость раствора ВМС снижается и при определенных значениях градиента скорости надмолекулярные структуры могут разрушаться, вследствие чего раствор приобретет свойства ньютоновской жидкости. Жидкости, проявляющие подобные эффекты ориентации, называют неньютоновскими. Вязкость растворов, содержащих макромолекулы полимера, обычно значительно выше вязкости растворов низкомолекулярных соединений и коллоидных растворов тех же концентраций. Поэтому только очень разбавленные растворы ВМС в условиях ламинарного течения можно считать ньютоновскими.
Увеличение вязкости раствора полимера по сравнению с вязкостью растворителя обусловлено не только его концентрацией, но и рядом параметров макромолекулы. Такими параметрами являются объем раствора, занимаемый макромолекулой (удельный объем), отношение длины молекулы к ее ширине (осевое отношение), а также жесткость молекулы. Для глобулярных молекул, каковыми являются молекулы многих белков, принципиальное значение имеет молекулярный объем. Его можно легко связать с относительной молекулярной массой. В случае очень жестких тонких молекул, как, например, ДНК, основной эффект оказывает осевое отношение; оно также является функцией относительной молекулярной массы. Если же известна относительная молекулярная масса, то можно получить информацию об общей форме молекулы.
Поскольку измерения абсолютной вязкости затруднены, чаще определяют относительную вязкость. При добавлении полимера к растворителю с вязкостью h0 вязкость раствора увеличивается до h. Отношение вязкости раствора к вязкости чистого растворителя называется относительной вязкостью hотн.: hотн. =  (33) Относительное повышение вязкости раствора ВМС по сравнению с вязкостью растворителя называется удельной вязкостью (hуд. ) и она равна:
(33) Относительное повышение вязкости раствора ВМС по сравнению с вязкостью растворителя называется удельной вязкостью (hуд. ) и она равна:
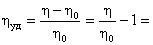 hотн. – 1 (34)
hотн. – 1 (34)
Относительная и удельная вязкости являются безразмерными величинами и зависят от концентрации полимера, а также градиента скорости. Но их невозможно связать непосредственно с параметрами макромолекулы (например, с её формой и объемом), поэтому были введены понятия приведенной и характеристической вязкостей. Удельная вязкость, отнесенная к единице концентрации, называется приведенной вязкостью hприв.. Её рассчитывают по формуле:
 , (35)
, (35)
где С – массовая концентрация полимера, г/см 3 .
Предельное значение приведенной вязкости в бесконечно разбавленном растворе назвали внутренней или характеристической вязкостью [h]:
 , (36)
, (36)
Экспериментально ее определяют путем построения графика зависимости приведенной вязкости (hуд/С) от различных концентраций полимера (рис. 49).
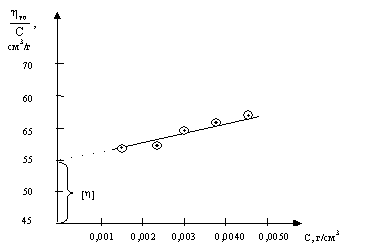
Рис.49. График зависимости приведенной вязкости от концентрации раствора ВМС
Такой график для достаточно разбавленных растворов полимеров носит прямолинейный характер. Экстраполируя прямую hуд./С = f(С) к С=0, на оси ординат отсекают отрезок, который соответствует предельному значению приведенной вязкости, т. е. характеристической вязкости [h].
Приведенная и характеристическая вязкости имеют размерности, обратные концентрации, т. е. см3/г.
Характеристическая вязкость характеризует гидродинамическое сопротивление потоку жидкости молекул полимера. Она зависит от относительной молекулярной массы, формы и удельного объема макромолекулы, её способности изменять форму в зависимости от природы растворителя (конформационные изменения), но она не зависит от концентрации полимера в растворе и скорости взаимного перемещения слоев жидкости.
Соотношение между характеристической вязкостью и относительной молекулярной массой полимера. Штаудингер предложил формулу для определения относительной молекулярной массы ВМС:
hуд. = КМ×С (37)
где hуд. – удельная вязкость раствора; К – константа, см3/г; С – концентрация ВМС в растворе, г/см3; М – относительная молекулярная масса ВМС.
Из уравнения (37) следует:  (38)
(38)
Иными словами, отношение удельной вязкости к концентрации полимера (т. е. приведенная вязкость) пропорциональна его относительной молекулярной массе и не зависит от его концентрации в растворе.
При выводе уравнения (37) Штаудингер допустил, что приведенная вязкость не зависит от концентрации полимера и что линейные макромолекулы в растворе ведут себя как жесткие стержни. Но на самом деле это не так. Были предложены многочисленные эмпирические формулы, в которых их авторы пытались устранить недостатки уравнения Штаудингера. Наиболее широкое применение нашло так называемое обобщенное уравнение Штаудингера или уравнение Марка-Хаувинка-Куна:
[h] = KM a, (38)
где К и a – постоянные для данного полимергомологического ряда и данного растворителя.
Эти константы обычно определяют опытным путем для каждой системы растворитель – растворенное вещество, используя соединения с известной относительной молекулярной массой, потому что до сих пор нет теории, пригодной для их расчета. Константы К и a, определенные для данной системы полимер – растворитель, нельзя использовать для другой системы.
Константа К имеет величину порядка 10–4. У жестких макромолекул
a » 1, для гибких полимерных молекул, приближающихся по форме к сфере,
a » 0,5, а у сильно заряженных полиэлектролитов a » 2.
Зависимость (38) можно записать также в виде:
ln[h] = ln K + alnM, (39)
Данное уравнение является уравнением прямой в координатах ln M, ln[h]. Измерив характеристическую вязкость нескольких стандартных препаратов с известными относительными молекулярными массами и разместив соответствующие точки в координатах lnM, ln[h] – рис.50, можно убедиться в справедливости выражения (39 ) для данного случая. Если нанесенные на график точки действительно лежат на одной прямой, то длина отрезка, отсекаемого ею на оси ln[h], и тангенс угла b ее наклона дают соответственно величины lnK и a в формуле (39). Теперь не составляет труда вычислить или определить непосредственно на графике неизвестную относительную молекулярную массу фракции полимера, для которой измерена характеристическая вязкость.

Рис.50.Зависимость характеристической вязкости от относительной
молекулярной массы фракций полимергомологического ряда
Вискозиметрия – это гидродинамический метод, основанный на измерении вязкости жидкостей и растворов. Метод позволяет определить относительную молекулярную массу растворенного полимера, а так же получить данные о размерах и форме его молекул. Вязкость можно определять различными способами, например методом падающего шарика, методом истечения жидкости через капилляр и др.
Определение вязкости методом истечения жидкости основано на измерении времени истечения одинаковых объемов раствора и растворителя через один и тот же капилляр и при одной и той же температуре, что позволяет рассчитать относительную вязкость. Согласно закону Пуазейля, объем жидкости V, перетекающей через капиллярную трубку, прямо пропорционален времени перетекания t, давлению столба жидкости р, четвертой степени радиуса капилляра r и обратно пропорционален длине капилляра и вязкости h:
 (40)
(40)
Из этого следует, что вязкость равна:
 (41)
(41)
 Для измерения вязкости данным методом чаще используют капиллярные вискозиметры, представляющие собой видоизмененные варианты вискозиметра Оствальда (рис.51). В широкое колено 1 прибора заливают жидкость, которую затем переводят в колено 2 выше метки 3. Жидкости дают свободно вытекать через капилляр 5, при этом отмечают по секундомеру время прохождения мениска жидкости от метки 3 до метки 4. Для данного вискозиметра длина капилляра
Для измерения вязкости данным методом чаще используют капиллярные вискозиметры, представляющие собой видоизмененные варианты вискозиметра Оствальда (рис.51). В широкое колено 1 прибора заливают жидкость, которую затем переводят в колено 2 выше метки 3. Жидкости дают свободно вытекать через капилляр 5, при этом отмечают по секундомеру время прохождения мениска жидкости от метки 3 до метки 4. Для данного вискозиметра длина капилляра  и ее радиус r, а также объем вытекающей жидкости V постоянны. Следовательно, их можно заменить константой к:
и ее радиус r, а также объем вытекающей жидкости V постоянны. Следовательно, их можно заменить константой к:
 (42)
(42)
Тогда уравнение (41) принимает вид:
h = к×pt, (43)
Согласно данному уравнению при постоянном давлении столба жидкости вязкость пропорциональна времени истечения. В таком случае относительная вязкость выражается следующим уравнением:

 (44)
(44)
Если жидкости вытекают под влиянием собственной тяжести при равных высотах столба жидкости, то отношение давлений можно заменить отношением плотностей. Поскольку при измерении вязкости разбавленных растворов ВМС плотности растворителя и раствора считают равными друг другу, то относительную вязкость рассчитывают по формуле:
hотн.  , (45)
, (45)
где t – время истечения разбавленного раствора ВМС; t0 – время истечения чистого растворителя.
Измерив время истечения растворителя и растворов с различными концентрациями полимера и рассчитав последовательно относительную (45), удельную (34) и приведенную (35) вязкости для этих растворов, строят график зависимости приведенной вязкости hуд./С от концентрации С. Прямую экстраполируют на ось ординат и находят значение [h]. Затем по уравнению (38) рассчитывают относительную молекулярную массу полимера.
Применение вискозиметрии для медико-биологических исследований. Величина характеристической вязкости позволяет определить как относительную молекулярную массу полимера, так же размеры и форму ее макромолекул. Например, если растворы белков характеризуются величинами [h], лежащими между 3,0 и 4,0 см3/г, то столь малое значение этих величин указывает на глобулярную, весьма компактную структуру этих белков, форма которых весьма незначительно отличается от сферы. Большие значения [h] указывают либо на высокую степень асимметричности этих белков, либо на большой объем, занимаемый этими белками в растворе.
Зависимость приведенной вязкости растворов биополимеров от их концентрации для макромолекул с разными значениями относительной молекулярной массы графически выражаются прямыми с разным наклоном, который тем меньше, чем меньше масса макромолекулы (рис.52).
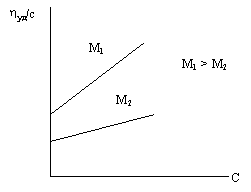
Рис.52. График зависимости отношения hуд/С от концентрации С
для двух разных молекул ДНК
Угол наклона прямых в этих же координатах зависит и от формы макромолекул. При одинаковых М для молекул со сферической симметрией прямая более пологая, чем для стержней.
При визкозиметрическом определении относительных молекулярных масс биополимеров используются разнообразные эмпирические формулы, связывающие [h] с М. Так, для белков, подвергнутых денатурации в 6М растворе хлорида гуанидиния, (вещество, которое разрывает все водородные связи так, что белок превращается в статистический клубок, если отсутствуют дисульфидные связи внутри одной полипептидной цепи) известно следующее соотношение:
[h]= 0,716 n0,66
где n - число аминокислотных остатков в белке. Зная среднюю молекулярную массу на один остаток, равную 115, и число аминокислотных остатков в белке, можно рассчитать его относительную молекулярную массу.
Для двухцепочечных линейных молекул ДНК было найдено, что соотношение между [h] и М можно записать следующим образом:
0,665 lgM = 2,863 + lg([h] + 5).
Это истинно эмпирическое уравнение можно использовать для вычисления М при условии, что образец ДНК гомогенен по молекулярной массе. Это ограничение следует иметь в виду вследствие большой чувствительности ДНК к деградации в процессе выделения и очистки.
Устойчивость растворов ВМС и способы выделения
биополимеров из их растворов
Растворы высокомолекулярных соединений – термодинамически устойчивые системы. Их устойчивость обусловлена не только хорошим сродством полимера с растворителем (лиофильностью), но в значительной степени конформационными возможностями полимерной цепи, то есть энтропийным фактором. Следовательно, нарушить устойчивость растворов полимеров возможно или уменьшением количества “свободного” растворителя или уменьшением энтропийного фактора. Первое достигается добавлением к раствору ВМС десольватирующих веществ, например, добавлением к водному раствору полимера – электролита. Понижение энтропийного фактора возможно за счет образования межмолекулярных связей, например, при увеличении концентрации полимера в растворе.
Высаливание. Процесс выделения ВМС из раствора при добавлении в раствор десольватирующих веществ (электролитов или неэлектролитов) называется высаливанием. Это явление не следует отождествлять с коагуляцией коллоидных систем. Например, коагуляция гидрозолей происходит при введении сравнительно небольших количеств электролита и представляет собой в основном необратимое явление. Высаливание же из водного раствора полимера происходит при добавлении относительно больших количеств электролита, не подчиняется правилу Шульце-Гарди и является вполне обратимым процессом – при добавлении избытка растворителя или после удаления из системы электролита (промыванием или диализом) высокомолекулярное вещество снова растворяется.
Различен и механизм обоих явлений. Коагуляция золей электролитами происходит в результате адсорбции ионов электролита и сжатия ДЭС коллоидных частиц при уменьшении агрегативной устойчивости системы. Выделение же ВМС из раствора при добавлении электролита объясняется уменьшением способности воды растворять полимер ввиду связывания её молекул гидратирующимися ионами электролита. Поэтому в первом случае достаточно незначительного количества электролита, а во втором – необходимо большое.
Добавление к системе с выпавшим ВМС растворителя (воды) восстанавливает сольватную (гидратную) оболочку полимера вследствие повышения количества “свободного” растворителя. Другими словами, растворителя хватает для восстановления сольватных (гидратных) оболочек макромолекул, а также для растворения полимера.
Из сказанного следует, что чем больше ион способен связывать растворитель, тем больше он будет уменьшать способность среды растворять высокомолекулярное вещество и, следовательно, высаливающее действие ионов соответствует их порядку в лиотропном ряду. Так катионы по мере уменьшения их высаливающего действия распологаются в ряд:
Li+ > Na+ > K+ > Rb+ > Cs+
Подобный же ряд анионов имеет вид:
SO42–> CH3COO– > Cl– > NO3– > Br– > I– > CNS–
Следует отметить, что обычно более сильный высаливающий эффект вызывают анионы.
Высаливание полимера путем добавления неэлектролитов принципиально не отличается от выделения ВМС из раствора электролитом. Обычно это жидкость, которая растворяет полимер хуже, чем растворитель. Например, для белка – это спирт, а для каучука – ацетон.
Высаливание является одним из методов фракционирования высокомолекулярных веществ, поскольку способность этих соединений выделяться из раствора весьма сильно зависит от их химической природы и резко возрастает с увеличением относительной молекулярной массы. Особенно широкое применение фракционирование с помощью высаливания приобрело для разделения белков. Чаще всего для высаливания белков используется сульфат аммония. Эта соль отличается хорошей растворимостью, мало изменяющейся при понижении температуры. Применяя водные растворы сульфата аммония разной концентрации, добиваются фракционированного осаждения белков: белки с большей относительной молекулярной массой осаждаются при добавлении растворов сульфата аммония малых концентраций и наоборот. При этом высаливание электролитом часто сочетают с введением в систему неэлектролита и охлаждением раствора.
Высаливание белков целесообразно проводить при значении pH среды, близком к изоэлектрической точке. При значениях рН больше или меньше ИЭТ возрастает заряд, вследствии чего молекулы растворителя активнее разрыхляют полимерную сетку, увеличивая устойчивость системы.
Застудневание. Ранее указывалось, что при ограниченном набухании образуется студень, который представляет собой пространственную сетку из макромолекул полимера, заполненную молекулами растворителя. Однако, может происходить и обратный процесс, когда раствор полимера переходит в состояние студня. Этот процесс называется застудневанием или желатинированием.
Сетчатые (пространственные) структуры формируются в студнях в результате возникновения водородных связей, электростатических взаимодействий или более прочных химических связей между различными участками макромолекул. Если эти связи в студне являются водородными или электростатическими, то прочность его мала и он легко разрушается. Примером таких систем служат студни желатины и агар-агара.
Процесс застудневания протекает в течение определенного промежутка времени не только при комнатной температуре, но и при более низких температурах. Время, необходимое для формирования рыхлых сетчатых структур студней, называется периодом созревания.
На процесс застудневания существенно влияют размеры и разветвленность макромолекул полимеров. Особенно легко образуют студни высокомолекулярные соединения, у которых длина макромолекул достигает несколько тысяч ангстрем и в тысячи раз превышает их поперечные размеры.
Более концентрированные растворы ВМС при прочих равных условиях легче дают студни, чем разбавленные. Например, растворы желатины с массовой долей ее 2% и более легко превращаются в студни при комнатной температуре. Растворы с меньшей массовой долей (0,5–1%) образуют не устойчивые студни, которые плохо сохранят форму; а еще более разбавленные не желатинируются вовсе. Зависимость процесса образования студня от концентрации объясняется тем, что в более концентрированных растворах уменьшается расстояние между макромолекулами и поэтому увеличивается число их столкновений и облегчается образование структур за счет их сцепления активными центрами.
Повышение температуры способствует усилению поступательного и колебательного движения макромолекул и благоприятствует разрыву связей между ними, что затрудняет застудневание. При понижении температуры ускоряется агрегация макромолекул полимера и процесс застудневания идет легче. Поэтому растворы, не застудневающие при комнатной температуре, в случае ее понижения образуют твердые студни.
Электролиты по-разному влияют на скорость застудневания: одни – ускоряют, другие – замедляют, а некоторые – даже исключают возможность перехода ВМС в студень. На застудневание главным образом влияют анионы. Экспериментально установлено, что соли серной и уксусной кислот ускоряют процесс застудневания, хлориды и иодиды замедляют, а роданиды приостанавливают его. По мере уменьшения действия анионов на процесс застудневания они располагаются в следующий ряд:
> CH3COO– > Cl– > N  > Br– > J– > CNS–
> Br– > J– > CNS–
Различия в указанных свойствах электролитов объясняются степенью их гидратации, которая уменьшается у анионов слева направо в этом ряду. Замедляющее действие анионов на процесс застудневания наблюдается, начиная с хлорид-иона.
Застудневание лучше всего протекает при рН раствора, соответствующем изоэлектрической точке белка.
Студни являются гомогенными системами, которые обладают упругими свойствами, нетекучи и способны сохранять форму. Упругость студней определяяется прочностью и гибкостью макромолекулярной сетки, а также свойствами ориентированных слоев молекул растворителя. Особенно характерно это для полярных макромолекул в водной среде. Гидратные оболочки, окружающие полярные группы, создают упругую водную сетку. Таким образом, жидкость, заполняющую сетку студня, можно условно разделить на две части: “свободную” и “связанную”, входящую в состав сольватных оболочек.
Связанная вода обладает особыми свойствами: большей плотностью, пониженной температурой замерзания (до –150), потерей растворяющей способности и т. д. Связанная вода студней играет большую роль в нашей жизни, по-скольку присутствие ее в почве, растениях, во всех живых организмах обеспечивает морозоустойчивость, поддерживает “водные запасы”, определяет морфологические структуры клеток и тканей.
При старении студни теряют гомогенность. Это явление называют синерезисом. Он сопровождается уплотнением пространственной структурной сетки и уменьшением объема студня за счет выделения жидкой фазы. Примеры синерезиса – отделение сыворотки при свертывании крови, при скисании молока и др. Студни не способны восстанавливать свою структуру.
Из-за наличия пространственной сетки в студнях отсутствует перемешивание. Поэтому в них реагирующие вещества соприкасаются в результате медленной диффузии и химические реакции имеют свои особенности, в частности, специфически протекают реакции осаждения. Например, если в студень желатины заранее ввести некоторое количество дихромата калия, а затем добавить более концентрированный раствор нитрата серебра, то возникает окрашенный осадок дихромата серебра:
K2Cr2O7 + 2AgNO3 ® Ag2Cr2O7¯ + 2KNO3
При стоянии в результате диффузии нитрата серебра осадок распространяется в глубь студня, но не сплошной массой: возникают периодические зоны осадка, отделенные друг от друга совершенно прозрачными промежутками. Эти реакции получили название периодических. Их впервые наблюдал немецкий химик Р. Лизенганг (1886).
Периодическими реакциями объясняют сложное распределение окраски многих минералов, генерацию нервных импульсов, мышечные сокращения, сложное строение камней, образующихся в почках, печени и желчном пузыре.
Коацервация. При нарушении устойчивости раствора белка или полисахарида возможно образование коацервата – новой жидкой фазы, обогащенной биополимером. Коацерват может выделяться в виде капель или образовывать сплошной слой, что приводит к расслаиванию системы на две фазы. Одна из фаз представляет собой раствор ВМС в растворителе, а другая – раствор растворителя в высокомолекулярном веществе.
Коацервацию можно вызвать изменением температуры, pH среды или введением низкомолекулярных веществ.
Наиболее изучена коацервация белков и полисахаридов в водных растворах. Л. И. Опарин считал, что коацерваты сыграли большую роль в процессах происхождения жизни на Земле.
Коацервацию используют при микрокапсулировании лекарственных веществ. Для этого лекарственное вещество диспергируют в растворе полимера. В результате на поверхности лекарственного вещества формируется оболочка из адсорбированных капелек коацервата полимера. Эти капельки сливаются в сплошной слой на поверхности частиц лекарственного вещества и специальной обработкой переводятся в твердое состояние. Образовавшаяся твердая оболочка обеспечивает устойчивость, увеличивает длительность действия и устраняет неприятный вкус лекарственного вещества.
Методы измерения ИЭТ белков
Свойства белков в изоэлектрической точке изменяются по сравнению с их обычным состоянием, что используется для измерения ИЭТ белка. В условиях ИЭТ вязкость растворов белков, их растворимость, степень гидратации и набухания становятся минимальными, а сами белки утрачивают электрофоретическую подвижностью. В ИЭТ белковые растворы подвергаются наибольшей коагуляции и имеют самую высокую скорость желатинирования.
Изоэлектрическую точку белков определяют прямыми и косвенными методами. К первым относятся методы, при которых определяется рН раствора белка, когда подвижность частичек в постоянном электрическом поле равна нулю (электрофоретические методы). Косвенные методы основаны на установлении рН раствора, при котором наблюдаются минимальные значения вязкости и степени набухания или максимальные значения скоростей желатинирования и коагуляции белка.
Пластификация биополимеров.
Пластификация — это введение в полимеры веществ (пластификаторов), повышающих эластичность и / или пластичность материала в условиях его эксплуатации и / или переработки.
Существует несколько методов пластификации:
Внешняя (первичная) пластификация — пластификация веществами, которые химически не связываются с полимером и могут удаляться путем испарения, экстракции и т.д.
Внутренняя (вторичная) пластификация — пластификация веществами, химически связывающимися с полимером, в результате чего свойства пластифицированных материалов стабильны во времени.
Внешняя пластификация, в свою очередь, делится на структурную и молекулярную. Молекулярный пластификатор — пластификатор первого рода — термодинамически совместим с полимером и действует подобно растворителю. Структурный пластификатор — пластификатор второго рода — термодинамически несовместим с полимером и действует на уровне крупных надмолекулярных структур.
Дата добавления: 2018-10-27; просмотров: 1446; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!