Пример реакции с участием фолиевой кислоты
Министерство здравоохранения ЛНР
ГУ «Луганский государственный медицинский университет им. Святителя Луки»
Кафедра медицинской химии
Контрольная работа
Вариант № 13
Выполнил: студент III курса
фармацевтического факультета
заочной формы обучения группы № 1
Молчановой В.В.
Проверил
Луганск – 2018
1. Витамин К: строение, суточная потребность, источники, биологическое значение. Проявления недостаточности витамина К в организме. Возможен ли гипервитаминоз витамина К, если да, то его причины и основные проявления?
Витамин К — общее название жирорастворимых термостабильных соединений, обладающих биологической активностью филлохинона; важны для образования нормальных количеств протромбина. Источники витамина. Витамин К синтезирует микрофлора кишечника. Дополнительные источники — листья люцерны, свиная печень, рыбная мука и растительные масла, шпинат, цветная капуста, плоды шиповника, зелёные томаты. Физиологическая роль витамина К — участие в активации факторов свёртывания крови (II [протромбин], VII [проконвертин], IX [фактор Кристмаса], X [фактор Стюарта—Прауэр]) в печени путём у-карбоксилирования остатков глутаминовой кислоты. Суточная потребность в витамине К — 0,2-0,4 мг. Недостаточность витамина К Недостаточность витамина К типична для новорождённых. У взрослых возникает на фоне основного заболевания. Причины недостаточности витамина К • Нарушение синтеза витамина К кишечной микрофлорой, например, при пероральном приёме антибиотиков и сульфаниламидов. • Нарушение всасывания витамина К. - Недостаточное поступление жёлчи — наружные билиарные свищи, обструкция желчевыводящих путей и т.д. - Употребление большого количества минеральных масел (например, вазелинового). • Нарушение функции печени (например, при гепатите, циррозе). • Лечение антикоагулянтами непрямого действия. • У новорождённых в возрасте от 3 до 5 сут кишечник ещё не заселён микрофлорой, способной синтезировать витамин К в достаточном количестве. Поэтому у детей первых дней жизни часто возникает геморрагический синдром. • Случаи дефицита витамина К вследствие недостаточного его поступления с пищей не описаны. Проявления недостаточности витамина К • Геморрагический синдром (носовые, желудочно-кишечные кровотечения, кровотечения из дёсен, внутрикожные и подкожные кровоизлияния), обычно сопровождающий основное заболевание. • При механической желтухе геморрагический синдром обычно появляется на 4-5-й день. • У новорождённых детей, находящихся на грудном вскармливании (грудное молоко содержит мало витамина К) и не получающих адекватные дозы витамина, могут возникать внутричерепные кровоизлияния или другие проявления геморрагического синдрома.
|
|
|
|
|
|
Диагноз недостаточности витамина К • Гипопротромбинемия ниже 30—35%, дефицит плазменных факторов VII, IX и X. • Протромбиновое время увеличено на 25% (патогномоничный признак дефицита витамина К при исключении другой патологии). • Дифференциальный диагноз — поражения печени, терапия антикоагулянтами или салицилатами; другие заболевания, сопровождающиеся кровоточивостью (цинга, аллергическая пурпура, лейкозы, тромбоцитопения). Лечение недостаточности витамина К • Диета. Включение в рацион продуктов, богатых витамином К (К1 — брюссельская и цветная капуста, шпинат, салат, кабачки; К2 — говяжья печень). • Фитоменадион 10 мг (доза для взрослых) п/к или в/м или при острой гипопротромбинемии в/в (в 5% растворе глюкозы или 0,9% растворе хлорида натрия со скоростью, не превышающей 1 мг/мин); внутрь по 5-20 мг 3—4 раза в сутки. Дозы и длительность лечения зависят от показателей свёртываемости крови (протромбиновый индекс, коагулограмма и др.). • Викасол 15—30 мг/сут внутрь или 10—15 мг/сут в/м. Профилактика • Послеоперационное назначение витамина К при парентеральном питании. • Новорождённым с целью профилактики геморрагической болезни рекомендовано назначение витамина К1 (фитоменадиона) 0,5-1 мг в/м или п/к сразу после рождения, при необходимости (например, если роженица получала лечение дифенином) инъекцию можно повторить через 6—8 ч. • Беременным не рекомендовано профилактическое применение перед родами ввиду возможного токсического влияния на плод. Гипервитаминоз К Гипервитаминоз К развивается только у новорождённых и характеризуется развитием гемолитического синдрома. Причины гипервитаминоза К • Применение препаратов витамина К (фитоменадиона, викасола) у детей с недостаточностью глюкозо-6-фосфатдегидрогеназы. • Передозировка препаратов витамина К. Проявления гипервитаминоза К У новорождённых большие дозы препаратов витамина К приводят к развитию гемолитической анемии, гипербилирубинемии и ядерной желтухи (особенно у недоношенных детей с эритробластозом). Профилактика гипервитаминоза К: назначение витамина К в адекватных дозах и своевременная диагностика ферментопатий.
|
|
|
2. Витамин Вс(фолиевая кислота): строение, суточная потребность, источники, биохимическое и биологическое значение, проявления недостаточности в организме.
|
|
|
Источники
Растительные продукты, дрожжи, мясо, печень, почки, желток яиц. Витамин активно синтезируется дружественной кишечной микрофлорой.
Суточная потребность
400 мкг.
Строение
Витамин представляет собой комплекс из трех составляющих – птеридина, пара-аминобензойной кислоты и глутаминовой кислоты. Остатков глутамата, соединенных через γ-карбоксильную группу, может быть разное количество.
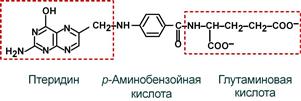
Строение фолиевой кислоты
Биохимические функции
Коферментной формой витамина является тетрагидрофолиевая кислота (ТГФК, Н4ФК).

Строение тетрагидрофолиевой кислоты
Непосредственная функция тетрагидрофолиевой кислоты – перенос одноуглеродных фрагментов, которые присоединяются к атомам N5 или N10:
· формила– в составе N5-формил-ТГФК и N10-формил-ТГФК,
· метенила– в качестве N5,N10-метенил-ТГФК,
· метилена– в виде N5,N10-метилен-ТГФК,
· метила– в форме N5-метил-ТГФК,
· формимина – в составе N5-формимино-ТГФК.

Строение и взаимопревращение активных форм
тетрагидрофолиевой кислоты
Благодаря способности переносить одноуглеродные фрагменты, витамин:
· участвует в синтезе пуриновых оснований и тимидинмонофосфата, и, следовательно, в синтезе ДНК,
· участвует в обмене аминокислот – обратимое превращение глицина и серина.
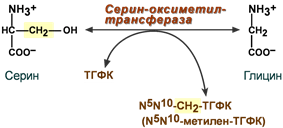
Пример реакции с участием фолиевой кислоты
· N5-метил-ТГФК взаимодействует с витамином В12, являясь донором метильной группы при превращении гомоцистеина в метионин.
В клетке N5-метил-ТГФК образуется в необратимой реакции из N5,N10-метилен-ТГФК. При этом единственнымспособом получить свободную ТГФК для других клеточных нужд является реакция превращения гомоцистеина в метионин. При дефиците витамина В12 эта реакция нарушается и возникает внутриклеточный дефицит витамина B9, хотя и в клетке и в крови его (в виде метил-ТГФК) может быть много. Такое явление получило название "ловушка для фолата".
Гиповитаминоз
Причина
Пищевая недостаточность, кислые продукты, тепловая обработка пищи, прием лекарств (барбитураты, сульфаниламиды и антибиотики, некоторые цитостатики – аминоптерин, метотрексат), алкоголизм и беременность.
Клиническая картина
В первую очередь затрагиваются органы кроветворения: так как клетки не теряют способности расти, но в них происходит нарушение синтеза ДНК с остановкой деления, то это приводит к образованию мегалобластов (крупных клеток) и мегалобластической анемии. Лейкопения присутствует по той же причине.
Аналогично развивается поражение слизистых желудка и кишечного тракта (гастриты, энтериты), глоссит.
Отмечается замедление роста, конъюнктивит, ухудшение заживления ран, иммунодефициты, оживление хронических инфекций и субфебрилитет.
Лекарственные формы
Фолинат кальция.
Антивитамины
Антивитамином В9 является группа лекарственных антибактериальных соединений сульфаниламидовструктурно схожих с компонентом фолиевой кислоты - парааминобензойной кислотой (ПАБК). В бактериальной клетке происходит конкуренция за активный центр фермента и нарушается использование ПАБК для синтеза фолиевой кислоты, что ведет к прекращению синтеза тимидилового нуклеотида, подавлению синтеза ДНК и размножения бактерии.

Сходство строения сульфаниламидов
и парааминобензойной кислоты
3. Охарактеризуйте механизм действия гормонов путем активации транспортных каналов. Для каких гормонов характерен? Его значение?
Гормоны, секретируемые железами внутренней секреции, связываются с транспортными белками плазмы или в некоторых случаях адсорбируются на клетках крови и доставляются к органам и тканям, влияя на их функцию и обмен веществ. Некоторые органы и ткани обладают очень высокой чувствительностью к гормонам, поэтому их называют органами-мишенямиилитканями–мишенями. Гормоны влияют буквально на все стороны обмена веществ, функции и структуры в организме.
Согласно современным представлениям, действие гормонов основано на стимуляции или угнетении каталитической функции определенных ферментов. Этот эффект достигается посредством активации или ингибирования уже имеющихся ферментов в клетках за счет ускорения их синтеза путём активации генов. Гормоны могут увеличивать или уменьшать проницаемость клеточных и субклеточных мембран для ферментов и других биологически активных веществ, благодаря чему облегчается или тормозится действие фермента.
Различают следующие типы механизма действия гормонов: мембранный, мембранно-внутриклеточный и внутриклеточный (цитозольный).
Мембранный механизм. Гормон связывается с клеточной мембраной и в месте связывания изменяет её проницаемость для глюкозы, аминокислот и некоторых ионов. В этом случае гормон выступает как эффектор транспортных средств мембраны. Такое действие оказывает инсулин, изменяя транспорт глюкозы. Но этот тип транспорта гормонов редко встречается в изолированном виде. Инсулин, например, обладает как мембранным, так и мембранно-внутриклеточным механизмом действия.
Мембранно-внутриклеточный механизм. По мембранно-внутриклеточному типу действуют гормоны, которые не проникают в клетку и поэтому влияют на обмен веществ через внутриклеточного химического посредника. К ним относят белково-пептидные гормоны (гормоны гипоталамуса, гипофиза, поджелудочной и паращитовидной желез, тиреокальцитонин щитовидной железы); производные аминокислот (гормоны мозгового слоя надпочечников – адреналин и норадреналин, щитовидной железы – тироксин, трийодтиронин).
Функции внутриклеточных химических посредников гормонов выполняют циклические нуклеотиды – циклический 3׳,5׳ –аденозинмонофосфат (цАМФ) и циклический 3׳,5׳ –гуанозинмонофосфат (цГМФ), ионы кальция.
Гормоны влияют на образование циклических нуклеотидов: цАМФ – через аденилатциклазу, цГМФ – через гуанилатциклазу.
Аденилатциклаза встроена в мембрану клетки и состоит из 3-х взаимосвязанных частей: рецепторной (R), представленной набором мембранных рецепторов, находящихся снаружи мембраны, сопрягающей (N), представленной особымN–белком, расположенным в липидном слое мембраны, и каталитической (C), являющейся ферментным белком, то есть собственно аденилатциклазой, которая превращает АТФ (аденозинтрифосфат) в цАМФ.
Аденилатциклаза работает по слудующей схеме. Как только гормон связывается с рецептором (R) и образуется комплекс гормон- рецептор, происходит образовагние комплексаN– белок – ГТФ (гуанозинтрифосфат), который активирует каталитическую (С) часть аденилатцеклазы. Активация аденилатциклазы приводит к образованию цАМФ внутри клетки на внутренней поверхности мембраны из АТФ.
Даже одна молекула гормона, связавшегося с рецептором, заставляет работать аденилатцеклазу. При этом на одну молекулу связавшегося гормона образуется 10-100 молекул цАМФ внутри клетки. В активном состоянии аденилатциклаза находится до тех пор, пока существует комплекс гормон – рецептор. Аналогичным образом работает и гуанилатциклаза.
В цитоплазме клетки находятся неактивные протеинкиназы. Циклические нуклеотиды- цАМФ ицГМФ- активируют пртеинкиназы. Существуют цАМФ- зависимые и цГМФ – зависимые протеинкиназы, которые активируются своим циклическим нуклеотидом. В зависимости от мембранного рецептора, связывающего определенный гормон, включается или аденилатцеклаза, или гуанилатцеклазаи соответственно происходит образование или цАМФ, или цГМФ.
Через цАМФ действует большинство гормонов, а через цГМФ- только окситоцин, тиреокальцитонин, инсулин и адреналин.
При помощи активированных протеинкиназ осуществляется два вида регуляции активности ферментов: активация уже имеющихся ферментов путем ковалентной модификации, то есть фосфолированием; изменение количества ферментного белка за счет изменения скорости его биосинтеза.
Влияние циклических нуклеотидов на биохимические процессы прекращается под влиянием специального фермента- фосфодиэстеразы, разрушающей цАМФ и цГМФ. Другой фермент – фосфопротеидфосфаза – разрушает результат действия протеинкиназы, то есть отщепляет фосфорную кислоту от ферментных белков, в результате чего они становятся неактивными.
Внутри клетки ионов кальция содержится очень мало, вне клетки их больше. Они пступают из внеклеточной среды по кальциевым каналам в мембране. В клетке кальций взаимодействует с кальцийсвязывающим белком калмодулином (КМ). Этот комплекс изменяет активность ферментов, что ведет к изменению физиологический функций клеток. Через ионы кальция действуют гормоны окситоцин, инсулин, простагландин F2α. Таким образом, чувствительность тканей и органов к гормонам зависит от мембранных рецепторов, а специфическое регуляторное влияние их определяется внутриклеточным посредником.
Внутриклеточный (цитозольный) механизм действия. Он характерен для стероидных гармонов (кортикостероидов, половых гормонов – андрогенов, эстрогенов и гестагенов). Стероидгные гормоны взаимодействуют с рецепторами, находящимися в цитоплазме. Образовавшийся гормнон-рецепторный комплекс переносится в ядро и действует непосредственно на геном, стимулируя или угнетая его активность, т.е. действует на синтез ДНК, изменяя скорость транскрипции и количество инфармационной (матричной) РНК (мРНК). Увеличение или уменьшение количества мРНК влияет на синтез белка в процессе трансляции, что приводит к изменению функциональной активности клетки.
4. Гормоны пучковой зоны коркового вещества надпочечников: их структура, особенности образования, механизм действия, биохимические эффекты, биологическая роль, патология при нарушении синтеза. Синдром Иценко-Кушинга.
Тотальный гиперкортицизм.Этиология и патогенез гиперкортицизма.Так как корковое вещество надпочечников представляет собой гормонообразующий комплекс из минералокортикоидов, глюкокортикоидов, андрогенов и поскольку стероидные гормоны частично перекрывают биологические эффекты друг друга, патология гиперкортицизма весьма мозаична. Функциональным регулятором для всех зон служит АКТГ (для пучковой зоны его роль безраздельная), и потому синдром тотального гиперкортицизма включает безусловную гиперпродукцию глюкокортикоидов нередко с более или менее выраженными симптомами гиперальдостеронизма и гиперандрогенизма.
По этиологии и патогенезу развития тотального гиперкортицизма выделяют следующие его варианты:
I.Первичный надпочечниковый гиперкортицизмкак результатпервичной гиперплазии железы (АКТГ-независимый) – синдром Иценко-Кушинга;
II.Вторичный гиперкортицизм при избыточный гипоталамо-гипофизарной стимуляции железы (АКТГ-зависимый) – болезнь Иценко-Кушинга;
III.Вторичный гиперкортицизм при избыточной эктопической продукции АКТГ вне гипоталамо-гипофизарной области;
IV. Ятрогенный гиперкортицизмпри экзогенном введении кортикостероидов.
I. В четверти случаев гиперкортицизм связан с первичным опухолевым поражением коркового вещества железы. Такая патология именуется АКТГ-независимым синдромом Иценко-Кушинга.Чаще всего эта опухоль растет из клеток пучковой зоны – глюкостерома (с избытком глюкокортикоидов). Разновидностью глюкостеромы является глюкоандростерома с избыточным синтезом в дополнение андрогенов. В этом случае картина синдрома Иценко-Кушинга сочетается с гиперандрогенизмом: у мальчиков в виде преждевременного полового созревания, у женщин – вирилизмом.
Другой причиной АКТГ-независимого синдрома Иценко-Кушинга является первичная двусторонняя неопухолевая гиперплазия коры надпочечников. Она встречается у подростков и молодых лиц. Ведущим звеном патогенеза признаётся аутоиммунный стимулирующий механизм, подобный болезни Базедова. Экспериментально получены стероидогенные и митозогенные (ростовые) иммуноглобулины к клеткам коры надпочечников. В отдельных случаях первичная двусторонняя неопухолевая гиперплазия рассматривается как наследственный аутосомно-доминантный вариант синдрома – симпотомокомплекс Карнея. Довольно редкой причиной первичного гиперкортицизма является билатеральная гиперплазия коры надпочечников.Механизмом этого расстройства считается стимулирующее АКТГ-подобное действие желудочно-ингибирующего пептида, синтезируемого гастроинтестинальными железами.
II. В подавляющем большинстве случаев причиной гиперкортицизма является опухоль передней доли гипофиза – базофильная аденома, или хромофобные опухоли, секретирующие в избытке АКТГ – адренокортикотропиномы. Такая патология в России называется болезнью Иценко-Кушинга. Ее патогенез связывают с мутацией G белка клеток гипофиза, имеющего сродство к кортиколиберину, в результате чего адренокортикотрофы приобретают чрезмерную активность к этому рилизинг-фактору гипоталамуса.
«Допотопные» способы лечения болезни Иценко-Кушинга методом резекции или экстирпации адреналовой железы при нераспознанных аденомах гипофиза приводили к бурному росту той же адренокортикотропиномы вследствие стимуляции опухолевых клеток аденогипофиза гипоталамическим кортиколиберином на фоне возникшего гипокортицизма, и болезнь Иценко-Кушинга сменялась на синдром Нельсона [объёмный опухолевый рост в черепе без признаков гиперкортицизма (если резецировались надпочечники)].
III. Относительно редкой причиной вторичного гиперкортицизма являются эктопические опухоли из клеток диффузной эндокринной системы (апудомы), секретирующие АКТГ, реже кортиколиберины. Данная патология возникает при бронхогенном раке лёгких, карциномах пищеварительного тракта, медуллярном раке щитовидной железы, опухолях островков Лангерганса, тимомах. Такая форма гиперкортицизма иногда сочетается с гиперсекрецией опухолевыми клетками и других биологически активных веществ – вазопрессина, окситоцина, гастрина и т.п. По сути, описанная патология является содержанием паранеопластического синдрома опухолевого роста. Уровень АКТГ при эктопической секреции превышает его при болезни Иценко-Кушинга.
IV. Ятрогенный гиперкортицизм возникает при длительном лечении средними или кратковременной терапии сверхвысокими дозами препаратов глюкокортикоидов.
Патогенезпроявлений тотального гиперкортицизмаопределяется избытком гормонов коры надпочечников как результат гиперплазии адренокортикоцитов.
Глюкокортикоиды – гормоны универсального метаболического цикла. Абсолютными стимуляторами их секреции является АКТГ, поэтому картина гиперкортицизма определяется эффектами и кортикостероидов, и самим АКТГ (например, одним из результатов действия АКТГ может быть гиперпигментация кожи), а также проопиомеланокортина и его производных. Сочетание с чертами гиперальдостеронизма объясняется как следствие стимуляции АКТГ, так и минералокортикоидным эффектом больших доз глюкокортикоидов. Напомним, что минералокортикоиды – важнейшие регуляторы калий-натриевого и водного баланса, а андрогены – регуляторы половых функций, стресса и процессов анаболизма.
Болезнь Иценко-Кушинга. Снижение дофаминовой активности и повышение тонуса серотонинергической системы ЦНС усиливают выработку кортиколиберина, АКТГ и далее кортизола (вторичный кортизолизм) вследствие нарушения механизмов «обратной связи». Гиперкортизолизм не оказывает тормозящего влияния на центральные нервные структуры. Для заболевания характерно не только и не столько повышение секреции АКТГ, сколько стимуляция выработки им гормонов надпочечников – кортизола, кортикостерона, альдостерона, андрогенов.
Нарушения гипоталамо-гипофизарных взаимоотношений сочетаются с изменениями секреции остальных тропных гормонов гипофиза – тормозится продукция СТГ, снижается содержание гонадотропинов и тиротропного гормона, но повышается секреция пролактина.
Клиника болезни Иценко-Кушинга определяется расстройством всех видов метаболизма, регулируемого стероидными гормонами надпочечников.
Нарушение белкового обмена в целом протекает под знаком катаболизма протеинов преимущественно в мышцах и мезенхимальных элементах (миоцитах, клетках кожи, соединительной ткани, костей, лимфоидных органах), а в печени и ЦНС даже преобладают анаболические процессы. По этой причине развивается миастения (мышечная слабость), гипотрофия мышц. Нарушение синтеза протеинов отражается на белковом составе соединительной ткани, гликозаминогликанов, содержании белка в плазме крови (особенно альбуминов), иммуноглобулинов (антител). Усиленное дезаминирование аминокислот приводит к гиперазотурии. Угнетается коллагеногенез, что приводит к истончению и растяжению кожи в местах скопления жира (симптом папиросной бумаги), способствующих образованию характерных стрий (полосы растяжения) багрово-фиолетового цвета вследствие вазопатий, эритроцитоза и гипертензии. У молодых больных нарушаются рост и обмен витамина D. Тормозится заживление ран.
Жировой обмен.Самымхарактерным проявлением гиперкортицизма является ожирение центральной локализации: на фоне гипотрофии конечностей жир откладывается в области живота, лица, шеи, в межлопаточном пространстве. Наиболее вероятными причинами ожирения являются полифагия, гиперинсулинизм, неравномерное распределение инсулиновых и глюкокортикодных рецепторов в различных липоцитах, стимуляция кортикостероидами выработки адипоцитами лептина, прямые липогенетические эффекты АКТГ и глюкокортикоидов. Избыток глюкокортикоидных рецепторов наблюдается в центральных липоцитах, а инсулинизм усиливает в них липогенез, увеличивает поступление глюкозы и жирных кислот.
Избыток глюкокортикоидов оказывает липолитический эффект, вызывая гиперлипопротеинемию преимущественно II-типа (за счет липопротеидов низкой и очень низкой плотности, холестерина, триглицеридов), которая по механизму развития может быть отнесена к продукционной и ретенционной формам. Развитие гиперлипопротеинемии связывают с усиленным синтезом в печени триглицеридов, липолизом, блокированием апо-B рецепторов многих клеток потребителей.
Углеводный обмен. Глюкокортикоиды оказывают контринсулярное действие – они тормозят работу переносчиков глюкозы (глюты-4) в инсулинзависимые ткани (липоциты, миоциты, клетки иммунной системы) в пользу инсулиннезависимых органов – ЦНС, сердца, диафрагмы и других. В печени усиливаются глюконеогенез, глюкогенез, гликогенез. У части больных с недостаточными резервами β-клеток pancreas формируется вторичный инсулиннезависимый сахарный диабет, осложняемый кетоацидозом вследствие высокой кетогенности глюкокортикоидов (что, кстати, характерно для инсулинзависмого сахарного диабета). У других больных в случае гиперфункции β-клеток островков Лангерганса развивается гиперинсулинизм, который стабилизирует ситуацию, и явного стероидного диабета не наступает.
Водно-солевой обмен и кислотно-основной баланс. Характеризуются задержкой натрия и потерей ионов водорода и калия, из-за чего содержание K+ в клетках возбудимых тканей (нейроны, кардиомиоциты, миоциты), а также в плазме крови и эритроцитах значительно снижается. Развивается гипокалиемический алкалоз. Растут объемы внеклеточной жидкости и крови (гиперволемия, плетора). Тормозится всасывание кальция в кишечнике, а в почках его экскреция усиливается. Развиваются нефрокальциноз и нефролитиаз, присоединяется вторичный пиелонефрит. Результатом может быть почечная недостаточность. Снижение кальция в организме приводит к развитию вторичного гиперпаратироидизма. Паратгормон активизирует переход стволовых костных клеток в остеокласты и тормозит превращение последних в остеобласты. Кортизол также ингибирует переход остеокластов в остеобласты. Увеличение остеокластов и повышение их активности вызывают резорбцию костной ткани. Последняя теряет способность фиксировать кальций, вследствие чего формируется остеопороз.
Сердечно-сосудистая система. Хронический гиперкортицизм вызывает симптоматическую гипертензию, развитие которой связывают со следующими механизмами:
1) увеличение объема крови (гиперволемия, плетора),
2) повышение чувствительности адренорецепторов резистивных сосудов к прессорным факторам из-за нарастания содержания натрия и снижения калия в миоцитах резистивных сосудов (то есть вследствие повышения их вазомоторного тонуса),
3) отек гладкой мускулатуры артериол и венул,
4) активация ренин-ангиотензиновой системы вследствие стимуляции глюкокортикоидами синтеза печенью α2-глобулина (ангиотензиногена) и эндотелина I,
5) ингибирующее влияние кортикостероидов на выделение предсердного натрийуретического пептида.
В иммунной системе формируются вторичный иммунодефицит, фагоцитарная недостаточность, проявляющиеся снижением устойчивости к инфекционным заболеваниям. Развиваются кожные бактериальные и грибковые инфекции. По этой причине и вследствие избытка андрогенов появляется угревая сыпь (acne vulgaris) и пустуло-папулезный околоротовой дерматит.
Половые функции. Одним из ранних и постоянных проявлений болезни Иценко-Кушинга является нарушение половой функции, которое вызвано снижением гонадотропной функции гипофиза и повышением секреции андрогенов корой надпочечников. У мужчин тормозится продукция андрогенов половыми железами (вследствие подавления секреции гонадолиберина и лютеинизирующего гормона по механизму контроля обратной связи), снижается либидо и развивается импотенция. Избыток андрогенов в гормональном наборе гиперкортицизма у женщин формируют гирсутизм (избыточное оволосение), маскулинизацию (приобретение мужского типа телосложения), изменение сексуального поведения, дисменорею, аменорею, спонтанные аборты, преждевременные роды, вторичное бесплодие, вирилизацию.
Нервная система. Острый избыток глюкокортикоидов индуцирует эйфорию, психоз, галлюцинации и мании, а хронический – депрессию.
Изменения в крови.Глюкокортикоиды стимулируют эритро- и лейкопоэз, запускают апоптоз лимфоцитов и эозинофилов, вследствие чего развиваются эритроцитоз, нейтрофилез, лимфопения, эозинопения, изменяют состояние свертывающей и антисвертывающей систем крови (развитие тромбогеморрагических синдромов).
Парциальный гиперкортицизм.Он обусловлен выраженнымпреобладанием секреции одной группы кортикостероидов над остальными и представлен следующими видами:
1) гиперальдостеронизм (первичный и вторичный);
2) адреногенитальный синдром (гиперандрогенизм).
В то же время чистых парциальных форм практически не бывает.
Первичный гиперальдостеронизм (синдром Конна).
I. Причиной являются опухоли клубочковой зоны (альдостерома) или с эктопической локализацией (яичник, кишечник, щитовидная железа). Избыток минералокортикоидов не ингибирует продукцию АКТГ в отличие от глюкостером, поэтому атрофии здоровой части надпочечников не наступает.
II. Доброкачественная наследственная глюкокортикоид-подавляемая альдостерома.
III. Билатеральная гиперплазия клубочковой зоны коры надпочечеников неизвестной этиологии. Как и в случае микронодулярной гиперплазии коры, в этиологии обсуждается роль стимулирующих антител.
IV. При употреблении в пищу корня солодки (лакрицы) и применении ее препаратов нарушается превращение кортизола в кортизон (наличие гиперризиниевой кислоты в растительном сырье тормозит фермент 11-β-гидроксилазу). В этом случае воспроизводится синдром псевдогиперальдостеронизма. Аналогичный дефект фермента является причиной гипертензивной формы наследственной гиперплазии коры надпочечников.
V. Синдром Лидля – псевдогиперальдостеронизм вследствие первичной рецепторной гиперчувствительности к альдостерону при нормальном его содержании в крови.
VI. Ятрогенное введение альдостерона.
При всех формах первичного гиперальдостеронизма продукция ренина в отличие от вторичных низкая. Гиперволемия через рецепторный механизм тормозит синтез ренина.
Вторичный гиперальдостеронизм.Развивается вследствие активации ренин-ангиотензин-альдостероновой системы и протекает с высоким уровнем ренина в плазме крови. Причинами вторичной избыточной секреции альдостерона являются:
1) Ишемия почек, вызванная поражением почечных артерий;
2) Гиповолемия;
3) Гипонатриемия и чрезмерная потеря натрия;
4) Первичная неопухолевая гиперплазия клеток юкстагломерулярного аппарата почки (синдром Барттера, избыток простагландинов Е2);
5) Рениномы (опухоли клеток юкстагломерулярного аппарата почки);
6) Беременность – эстрогены стимулируют синтез ренина и ангиотензиногена.
Патоморфология. При вторичном гиперальдостеронизме нет опухоли и узелковой гиперплазии, а наблюдается гиперсекреция и диффузная гипертрофия-гиперплазия.
Проявления гиперальдостеронизма складываются из типичных симптомов:
1) электролитно-водные нарушения – гипернатриемия и задержка воды (гиперволемия), гипокалиемия и потеря ионов водорода.
2) гипертензия. Она сопровождается ортостатическими колебаниями (из-за экскреции калия барорецепторы теряют чувствительность к изменениям систолического и диастолического артериального давления).
3) отсутствие отеков – компенсаторно усиливается продукция предсердных натрийуретических пептидов (атриопептиды). Этот механизм удаляет часть натрия и воды и тормозит образование отеков. Потери калия также сопровождаются полиурией главным образом в ночное время.
4) тяжелая гипокалиемия порождает мышечную слабость, нарушение поступления глюкозы с током калия в клетку (диабетогенное действие), «гипокалиемическую нефропатию» с полиурией.
5) алкалоз – сдвиг кислотно-основного равновесия в щелочную сторону (в дистальных извитых канальцах реабсорбция Na+ идет в обмен на выделение К+ и H+) сопровождается гипокальциемией с возможной тетанией.
Основным звеном патогенезавторичного гиперальдостеронизма является очень высокая активность ренин-ангиотензин-альдостероновой системы, которая протекает с выраженной гиперренинемией и гиперангиотензинемией, которые находятся в антагонистических отношениях с натрийуретическими пептидами. Поэтому формируется очень высокая гипернатриемия и системные отеки.
Адреногенитальный синдром.Он рассматривается как парциальная чрезмерная секреция в надпочечниках половых гормонов (гиперандрогенизм).
Нарушения продукции половых гормонов надпочечников является причиной сексуальных нарушений, получившие собирательное название – адреногенитальный синдром. К ним относят:
1. Приобретенные формы, связанные с различными опухолями:
1. болезнь и синдром Иценко-Кушинга, в том числе глюкоандростерома,
2. андростеромы,
3. кортикоэстромы (описаны отдельные случаи у мужчин).
2. Врожденные формы. Они входят в структуру адреногенитального синдрома под названием «врожденный адреногенитальный синдром»или «врожденная гиперплазия (дисплазия) коры надпочечников»(ВДКН). Причиной является многообразие генных мутаций, блокирующих разные этапы генетически детерминированного стероидогенеза.
Патогенез. Типичны симптомы женского гиперандрогенизма: гирсутизм, дисменорея, вирилизм и угри. У детей опухоль приводит к раннему половому созреванию. Рост детей останавливается. У девочек врожденный синдром протекает по гетеросексуальному типу и формирует псевдогермафродитизм, у мальчиков – по изосексуальному типу. В 75% случаев гипокортицизм проявляется и сопровождается врожденной гиперпигментацией кожи, потерей соли с мочой (полиурия, гипонатриемия, гипотония мышц, гиперкалиемия, гипохлоремия, ацидоз, гипотензия), рвотой фонтаном, тягой к соленой пищи. В 25% случаев гипокортицизм носит скрытый характер.
У женщин формируется вирилизм: гирсутизм, маскулинизация телосложения, перераспределение жира по мужскому типу, грубый голос, облысение, атрофия молочных желез, олигоменорея и аменорея, гипертрофия клитора, физическая выносливость, изменение стереотипов полового поведения. У мужчин такие опухоли остаются не распознанными. У них хорошо распознаются кортикоэстеромы – злокачественные образования с мутантной продукцией эстрогенов, которые вызывают феминизацию – гинекомастию, женский тип телосложения и поведения, гипотрофию тестикул. Наибольшего внимания требуют врожденные формы адреногенитального синдрома с метаболическим блоком синтеза кортизола в направлении андрогенов. Таких наследственных причин много. Они требуют дифферециально-диагностических различий от истинного и ложного гермафродитизма вненадпочечниковых и неэндокринных причин и определения хромосомного пола. Надпочечниковые врожденные формы гиперандрогенизма (адреногенитальный синдром) могут протекать в составе синдрома гипокортицизма при явлениях дефицита глюко- и минералокортикоидов.
Известны классические формы гиперандрогенизма: вирилизирующие плюс сольтеряющие и тольковирилизирующие. Неклассическая форма характеризуется поздним началом болезни.
Ведущим звеном патогенез является ферментативный блок превращения 17-оксипрогестерона в 11-дезоксикортизол, что ведет к избыточному превращению метаболитов в андростендион. Гиперандрогения развивается внутриутробно. Одновременно формируется дефицит синтеза минерало- и глюкокартикоидов. На этом фоне по механизму обратной связи растет секреция АКТГ и стимулируется рост коры надпочечников и андростероидогенез. Кора надпочечников увеличивается за счет клубочковой и сетчатой зон и напоминает кору больших полушарий мозга. Клинически адреногенитальный синдром складывается из двух синдромов – гиперандрогенизма и гипокортицизма, причем преимущественно в форме гипоальдостеронизма.
Стертые и легкие формы [«врожденная гиперплазия (дисплазия) коры надпочечников»] встречаются до 30%. Они являются причиной гирсутизма и адренархе. Гирсутизм является убедительным поводом для поиска синдрома дефекта 21-гидроксилазы. Дефект других ферментов стероидогенеза, создающих врожденную картину адреногенитального синдрома, встречается крайне редко и приводится в специальных руководствах.
4. Общая хар-ка гликозаминогликанов в основном веществе соединительной ткани, их строение, роль.
Соединительная ткань (СТ) составляет более 50% массы тела, является составной частью всех органов и систем, формируя вместе с кровью и лимфой внутреннюю среду организма. Широкое распространение СТ в организме определяет её важнейшую характеристику – универсальность. В её состав входят:
1. клеточные элементы – а) фибробласты, б) макрофаги (гистиоциты), в) тучные клетки (лаброциты). Гранулоциты, лимфоциты, плазмоциты проникают в СТ из крови;
2.
внеклеточный матрикс (ВМ), включающий: а) волокнистые структуры (коллагеновые, эластические, ретикулярные волокна) и б) основное вещество – аморфный компонент, в который погружены клетки и волокна, содержащий углеводно-белковые комплексы – протеогликаны (соединения гликозаминогликанов с белками) и гликопротеиды. (Таблица 1).
Эти элементы в различных количественных соотношениях обнаруживаются во всех разновидностях СТ.
Количество СТ в разных органах широко варьирует: кожа и кости состоят в основном из СТ, а в головном и спинном мозгу ее почти нет.
Биосинтез основных макромолекул СТ происходит внутри ее клеток. Только после выхода этих макромолекул во внеклеточное пространство между ними возникают взаимодействия – образуются протеогликаны, а также комплексы между коллагеном, протеогликанами и коллагеновыми волокнами.
Основными низкомолекулярными компонентами СТ являются вода и ионы натрия. Последние нейтрализуют отрицательный заряд гликозаминогликанов, а основная часть воды в СТ образует гидратную оболочку ионов.
Разнообразие компонентов СТ и их особенностей обуславливают еще один признак ее – гетерогенность.
Общность происхождения элементов СТ и выполняемых ими функций определяют СТ как единую функциональную систему, все элементы которой находятся в тесной взаимосвязи и взаимозависимости. В связи с этим изменения любого компонента этой системы неизбежно отражается на других и, в конце концов, приводят к изменению всей системы в целом.
Дата добавления: 2018-06-01; просмотров: 378; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!