Тройной тест второго триместра беременности
Тема №4. Наследственные болезни человека
Наследственные болезни - заболевания, возникновение и развитие которых связано с различными дефектами и нарушениями в наследственном аппарате клеток.
В результате изучения клиники (симптомов) наследственных болезней вы научитесь определять проблемы пациента (актуальные) и профилактировать развитие потенциальных проблем пациента.
В основе наследственных заболеваний лежат мутации: хромосомные, генные и митохондриальные.
Митохондриальные заболевания обусловлены генетическими, структурными, биохимическими дефектами митохондрий, приводящими к нарушениям тканевого дыхания. Они передаются только по женской линии к детям обоих полов, так как сперматозоиды передают зиготе половину ядерного генома, а яйцеклетка поставляет и вторую половину генома, и митохондрии.
Можно выделить две группы митохондриальных заболеваний:
Ярко выраженные наследственные синдромы, обусловленные мутациями генов, ответственных за митохондриальные белки (синдром Барта, синдром Кернса-Сейра, синдром Пирсона).
Вторичные митохондриальные заболевания, включающие нарушение клеточного энергообмена (болезни соединительной ткани, синдром хронической усталости, гликогеноз, кардиомиопатия, мигрень, печёночная недостаточность, панцитопения, а также гипопаратиреоз, диабет, рахит).
Рассмотрим синдром хронической усталости, как наиболее часто встречающееся в практике медицинского специалиста заболевание.
|
|
|
Симптомы:
1. отсутствие ощущения отдыха после полноценного ночного сна;
2. часто повторяющиеся головные боли без видимых на это причин;
3. повышенная сонливость в дневное время суток;
4. невозможность быстро заснуть даже после напряжённого физического труда;
5. немотивированное раздражение;
6. плохое настроение, поводов которому нет;
7. частые инфекционные заболевания;
8. аллергические реакции;
9. снижение памяти и способности концентрироваться;
10. фарингит;
11. воспалённые лимфатические узлы на шее и в подмышечной области;
12. необъяснимая мышечная боль.
Классификация наследственных болезней
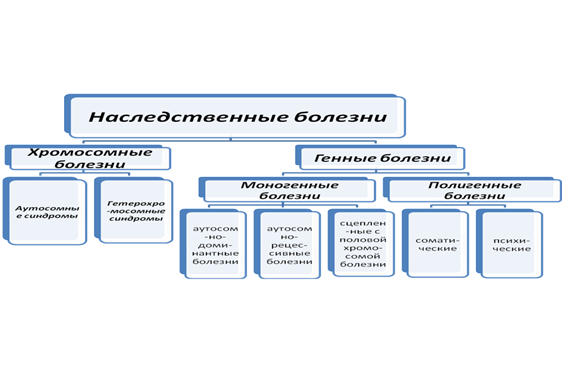
Хромосомные болезни развиваются в результате поломки наследственного материала на уровне хромосом.
Виды хромосомных поломок.
Качественные поломки:
1. делеция – потеря участка хромосомы в виде концевого отрыва или выпадения срединного участка хромосомы
2. транслокация- перенос оторвавшегося участка с одной хромосомы на другую
3. инверсия – поворот участка внутри хромосомы на 180°
4. дупликация – удвоение участка хромосомы
Количественные поломки:
|
|
|
1. Моносомия – отсутствие в хромосомной паре одной из хромосом (отцовской или материнской).
2. Полисомия – наличие в хромосомной паре лишней (добавочной) хромосомы, полученной от матери или отца.
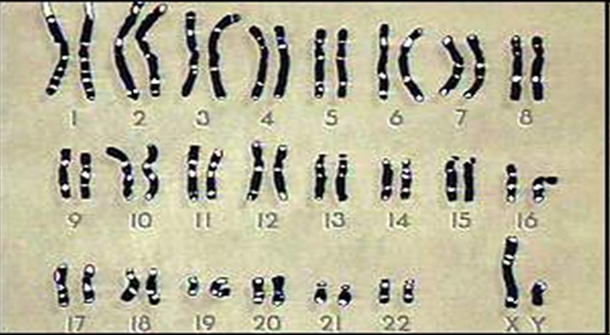
Нормальная хромосомная карта человека
Аутосомно-хромосомные синдромы
Синдром Патау – трисомия 13
История
Трисомия 13 впервые описана Эразмусом Бартолином в 1657 году. Хромосомную природу заболевания выявил доктор Клаус Патау в 1960 году. Заболевание названо в его честь.
Этиология и эпидемиология
Встречается с частотой 1:7000-1:14000.
Соотношение полов при синдроме Патау близко к 1:1. Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией (на 25 — 30 % ниже средних величин), которую нельзя объяснить небольшой недоношенностью (средний срок беременности 38,5 недель).
Проявления заболевания
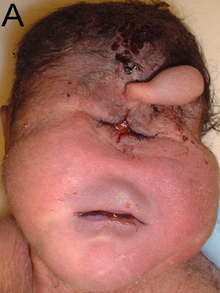
Младенец с синдромом Патау. Аринэнцефалия с циклопией
Характерным осложнением беременности при вынашивании плода с синдромом Патау является многоводие: оно встречается почти в 50 % случаев Синдрома Патау.
При синдроме Патау наблюдаются тяжёлые врождённые пороки. Дети с синдромом Патау рождаются с массой тела ниже нормы (2500 г). У них выявляются умеренная микроцефалия, нарушение развития различных отделов ЦНС, низкий скошенный лоб, суженные глазные щели, расстояние между которыми уменьшено, микрофтальмия и колобома, помутнение роговицы, запавшая переносица, широкое основание носа, деформированные ушные раковины, расщелина верхней губы и нёба, полидактилия, флексорное положение кистей, короткая шея. У 80 % новорождённых встречаются пороки развития сердца: дефекты межжелудочковой и межпредсердной перегородок, транспозиции сосудов и др. Наблюдаются фиброкистозные изменения поджелудочной железы, добавочные селезёнки, эмбриональная пупочная грыжа. Почки увеличены, имеют повышенную дольчатость и кисты в корковом слое, выявляются пороки развития половых органов. Для СП характерна задержка умственного развития.
|
|
|
В связи с тяжёлыми врождёнными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы (95 % — до 1 года).
Однако некоторые больные живут в течение нескольких лет. Более того, в развитых странах отмечаются тенденция увеличения продолжительности жизни больных синдромом Патау до 5 лет (около 15 % детей) и даже до 10 лет (2 — 3 % детей).
Оставшиеся в живых страдают глубокой идиотией.
|
|
|
Другие синдромы врождённых пороков развития (синдромы Меккеля и Мора, тригоноцефалия Опитца) по отдельным признакам совпадают с синдромом Патау. Решающим фактором в диагностике является исследование хромосом. Цитогенетическое исследование показано во всех случаях, в том числе у умерших детей. Точный цитогенетический диагноз необходим для прогноза здоровья будущих детей.
Лечение
Исправить хромосомные нарушения невозможно. Комплексная работа группы различных специалистов заключается в постоянном контроле за состоянием здоровья больного и поддержке семьи.
Синдром Э́двардса (синдром трисомии 18) — хромосомное заболевание, характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы. Описан в 1960 году Джоном Эдвардсом (John H. Edwards). Популяционная частота примерно 1:3000 в США, и 1:5000 в мире на 2016 год. Дети с трисомией в 18 хромосоме чаще рождаются у пожилых матерей. Для женщин старше 45 лет риск родить больного ребёнка составляет 0,7 %. Девочки с синдромом Эдвардса рождаются в три раза чаще мальчиков. Выживание после года жизни составляет около 5–10%.
Причины заболевания
Причиной заболевания является наличие дополнительной 18-й хромосомы (трёх вместо двух в норме для диплоидного набора) в кариотипе зиготы.
Симптомы
Дети с трисомией 18 рождаются с низким весом, в среднем около 2200 грамм, при этом длительность беременности — нормальная или даже превышает норму. Чаще всего возникают аномалии мозгового и лицевого черепа.
•Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие.
•Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует.
•Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной.
•В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщён и укорочен.
•Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов.
•У всех больных наблюдаются гипоплазия мозжечка, выраженная умственная отсталость, снижение мышечного тонуса.
Прогноз
Продолжительность жизни детей с синдромом Эдвардса невелика: 60 % детей умирают в возрасте до 3 месяцев, до года доживает лишь 5-10 %. Основной причиной смерти служат остановка дыхания и нарушения работы сердца. Оставшиеся в живых — глубокие олигофрены.
Частота появления
Частота появления синдрома Эдвардса составляет ~ 1:7000 зачатий и 1:8000 рождений живых детей. Риск рождения больного ребёнка увеличивается с возрастом, особенно, если мать болеет диабетом.
Синдром кошачьего крика (синдром Лежёна – делеция 5) — по имени описавшего его в 1963 году французского учёного) — редкое генетическое расстройство, вызываемое отсутствием фрагмента 5-й хромосомы.
Эпидемиология
Частота проявления синдрома примерно 1 : 45 000. Соотношение полов М1 : Ж1,3.
Генетика
Кариотип 46 XX или XY, 5р-. Диагноз подтверждается кариологическим исследованием с применением одного из методов идентификации хромосом.
Хромосомно синдром кошачьего крика развивается при делеции (с утратой от трети до половины, реже полная утрата) короткого плеча пятой хромосомы.
Клиника
При этом синдроме наблюдается:
· общее отставание в развитии,
· низкая масса при рождении и мышечная гипотония,
· лунообразное лицо с широко расставленными глазами,
· характерный плач ребёнка, напоминающий кошачье мяуканье, причиной которого является изменение гортани (сужение, мягкость хрящей, уменьшение надгортанника, необычная складчатость слизистой оболочки) или недоразвитие гортани. Признак исчезает к концу первого года жизни.
Кроме того, встречаются врождённые пороки сердца, костно-мышечной системы и внутренних органов, микроцефалия, птоз, низкое расположение и деформация ушных раковин, кожные складки впереди уха, гипертелоризм (увеличенное расстояние между какими-либо парными органами или анатомическими образованиями — например, между внутренними краями глазниц, грудными сосками), эпикантус (вертикальная кожная складка около внутреннего угла глаза, обычно двусторонняя; наиболее чётко выражена при синдроме Дауна), антимонголоидный разрез глаз.
Клиническая картина синдрома и продолжительность жизни людей с этим синдромом довольно сильно варьируются по сочетанию врождённых пороков развития органов.
Лечение
Лечение симптоматическое. Показаны средства, стимулирующие психомоторное развитие, лечебный массаж и гимнастика.
Синдром Уильямса (синдром «лица эльфа») делеция 7 — синдром, возникающий как следствие наследственной хромосомной перестройки. Люди, страдающие этим синдромом, обладают специфической внешностью и характеризуются общей задержкой умственного развития при развитости некоторых областей интеллекта.
Синдром описан в 1961 году кардиологом из Новой Зеландии Дж. Уильямсом, который выделил среди своих пациентов, у которых были обнаружены сходные дефекты сердечно-сосудистой системы, людей, которые также имели схожую внешность и умеренную отсталость.
Особенности внешности

Эльфы из английского фольклора кажутся детьми с синдромом Уильямса
Больные имеют особое строение лица, в специальной литературе называемое «лицом эльфа», поскольку оно напоминает лицо эльфов в их традиционном, фольклорном варианте. Для них характерны широкий лоб, разлёт бровей по средней линии, опущенные вниз полные щёки, большой рот с полными губами (особенно нижней), плоское переносье, своеобразная форма носа с плоским тупым концом, маленький, несколько заострённый подбородок.
Глаза зачастую ярко-голубые, со звёздчатой картиной радужки и склерами синеватого цвета. Разрез глаз своеобразный, с припухлостями вокруг век. Сходящееся косоглазие.
Для старших детей характерны длинные, редкие зубы. С возрастом лицо больных несколько меняется: появляется массивность надбровных дуг, меньше выражена пастозность лица, нет плоского переносья и эпиканта. Обращает на себя внимание увеличенное расстояние от основания носа до верхней губы.
Сходство лиц усиливает улыбка, которая ещё больше подчёркивает отёчность век и своеобразное строение рта.
Ни одна из этих черт не является обязательной, но их общее сочетание всегда присутствует
Психологические особенности
Умственная отсталость при синдроме Вильямса наблюдается во всех случаях. Для этого синдрома характерен дефицит наглядно-образного мышления, абстрактное мышление практически полностью отсутствует. Степень интеллектуального дефекта довольно значительна, IQ в среднем колеблется от 40 до 50 (умеренная умственная отсталость — имбецильность.
Можно отметить большое сходство психопатологической картины дефекта у всех больных: при значительном снижении интеллекта больные имеют относительно большой словарный запас, очень словоохотливы, склонны к подражанию, речь у детей довольно хорошая, тем не менее, она представляет всего лишь набор штампов. Вместе с тем всегда страдают пространственные представления, организация и планирование деятельности. Очень характерны и постоянны особенности личности этих детей: добродушие, приветливость, послушание, эмоциональность, стремление к общению, доверчивость. Практически всегда имеется хороший музыкальный слух даже при выраженном интеллектуальном дефекте. Нередко выявляются неврозоподобные нарушения — энурез, привычная рвота, навязчивые действия, головная боль. У части детей наблюдается гиперактивность, страхи и тревога.
Некоторые дети могут учиться во вспомогательной школе, они овладевают чтением и письмом, но им недоступны действия, связанные с организацией даже простейших трудовых операций.
Частота
Встречается с частотой приблизительно 1 на 20 000 новорожденных.
Лечение - симптоматическое
Синдро́м Да́уна (трисомия по хромосоме 21) — одна из форм геномной патологии, при которой чаще всего кариотип представлен 47 хромосомами вместо нормальных 46, поскольку хромосомы 21-й пары, вместо нормальных двух, представлены тремя копиями (см. также плоидность). Существует ещё две формы данного синдрома: транслокация хромосомы 21 на другие хромосомы (чаще — на 15, реже — на 14, ещё реже — на 21, 22 и Y-хромосому) — 4 % случаев, и мозаичный вариант синдрома — 5 %.
Синдром получил название в честь английского врача Джона Дауна, впервые описавшего его в 1866 году. Связь между происхождением врождённого синдрома и изменением количества хромосом была выявлена только в 1959 году французским генетиком Жеромом Леженом.
Первый Международный день человека с синдромом Дауна был проведён 21 марта 2006 года по инициативе греческого генетика Стилианоса Антонаракиса из Женевского университета. День и месяц были выбраны в соответствии с номером пары и количеством хромосом.
Эпидемиология

График показывает вероятность возникновения синдрома Дауна в зависимости от возраста матери
Синдром Дауна не является редкой патологией — в среднем наблюдается один случай на 700 родов; в данный момент благодаря пренатальной диагностике частота рождения детей с синдромом Дауна уменьшилась до 1 к 1100, так как узнавшие о заболевании плода прибегают к абортам. У обоих полов аномалия встречается с одинаковой частотой.
Частота рождений детей с синдромом Дауна 1 на 800 или 1000. В 2006 году Центр по контролю и профилактике заболеваний оценил её как один на 733 живорождённых в США (5429 новых случаев в год). Около 95 % из них по трисомии 21-й хромосомы. Синдром Дауна встречается во всех этнических группах и среди всех экономических классов.
Возраст матери влияет на шансы зачатия ребёнка с синдромом Дауна. Если матери от 20 до 24 лет, вероятность этого 1 к 1562, до 30 лет — 1 к 1000, от 35 до 39 лет — 1 к 214, а в возрасте старше 45, вероятность 1 к 19. Хоть вероятность и увеличивается с возрастом матери, 80 % детей с данным синдромом рождаются у женщин в возрасте до 35 лет. Это объясняется более высокой рождаемостью в данной возрастной группе. По последним данным отцовский возраст, особенно если старше 42 лет, также увеличивает риск синдрома.
Ожидаемая продолжительность жизни в развитых странах составляет от 50 до 60 лет при надлежащем медицинском обслуживании.
Пренатальная диагностика в России
В России беременные женщины в числе прочих исследований проходят скрининг на предмет вероятности рождения ребёнка с синдромом Дауна
Комбинированный тройной тест первого триместра беременности
При сроке беременности от 11 до 14 недель беременная женщина направляется в медицинскую организацию, осуществляющую экспертный уровень пренатальной диагностики, для проведения комплексной пренатальной (дородовой) диагностики нарушений развития ребёнка, включающей УЗИ врачами-специалистами, прошедшими специальную подготовку и имеющими допуск на проведение ультразвукового скринингового обследования в I триместре, и определение материнских сывороточных маркёров с последующим программным комплексным расчётом индивидуального риска рождения ребёнка с хромосомной патологией].
Расчёт риска производится по трём показателям, с учётом возраста женщины:
· количество ассоциированного с беременностью плазменного протеина А (pregnancy associated plasma protein-A, РАРР-А);
· количество свободной β-субъединицы хорионического гонадотропина человека (β-ХГЧ);
· ультразвуковые признаки (увеличение объёма жидкости в воротниковом пространствае, укорочение костей носа, укороченные костей голени, изменение структур мозга и другие).
Перечисленные методы не позволяют поставить точный диагноз, и в результате проведённого скрининга лишь формируется группа риска беременных с повышенной (индивидуальный риск 1/100 и выше) вероятностью рождения больного синдромом Дауна. На втором этапе в группе риска проводится инвазивная процедура для получения плодного материала, необходимого для точного проведения анализа на синдром Дауна. В зависимости от срока беременности это может быть биопсия ворсин хориона (8-12 недели), амниоцентез (14-18 недели) или кордоцентез (на более поздних сроках). В полученных образцах ткани плода проводится определение хромосомного набора.
В случае постановки диагноза хромосомных нарушений и врождённых аномалий (пороков развития) у плода с неблагоприятным прогнозом для жизни и здоровья ребёнка после рождения прерывание беременности по медицинским показаниям проводится независимо от срока беременности по решению перинатального консилиума врачей после получения информированного добровольного согласия беременной женщины.
Тройной тест второго триместра беременности
При сроке беременности от 16 до 18 недель производится биохимическое исследование исследование крови женщины, при котором оцениваются следующие показатели:
· количество α-фетопротеина (альфа-ФП);
· количество свободной β-субъединицы хорионического гормона человека (бета-ХГЧ);
· количество эстриола свободного.
При сроке беременности от 18 до 21 недели беременная женщина направляется в медицинскую организацию, осуществляющую пренатальную диагностику, в целях проведения УЗИ для исключения поздно манифестирующих врождённых аномалий развития плода.
В третьем триместре, при сроке беременности от 30 до 34 недель УЗИ проводится по месту наблюдения беременной женщины.
Постнатальная диагностика
симптомы
· «плоское лицо» — 90 %
· брахицефалия (аномальное укорочение черепа) — 81 %
· кожная складка на шее у новорожденных — 81 %
· эпикантус (вертикальная кожная складка, прикрывающая медиальный угол глазной щели) — 80 %
· гиперподвижность суставов — 80 %
· мышечная гипотония — 80 %
· плоский затылок — 78 %
· короткие конечности — 70 %
· брахимезофалангия (укорочение всех пальцев за счёт недоразвития средних фаланг) — 70 %
· катаракта в возрасте старше 8 лет — 66 %
· открытый рот (в связи с низким тонусом мышц и особым строением нёба) — 65 %
· зубные аномалии — 65 %
· клинодактилия 5-го пальца (искривлённый мизинец) — 60 %
· аркообразное нёбо — 58 %
· плоская переносица — 52 %
· бороздчатый язык — 50 %
· поперечная ладонная складка (называемая также «обезьяньей») — 45 %
· короткая широкая шея — 45 %
· ВПС (врождённый порок сердца) — 40 %
· короткий нос — 40 %
· страбизм (косоглазие) — 29 %
· деформация грудной клетки, килевидная или воронкообразная — 27 %
· пигментные пятна по краю радужки = пятна Брушфильда — 19 %
· эписиндром — 8 %
· стеноз или атрезия двенадцатиперстной кишки — 8 %
· врождённый лейкоз — 8 %.
Точная диагностика возможна на основании анализа крови на кариотип. На основании исключительно внешних признаков постановка диагноза невозможна.
Когнитивное развитие
Когнитивное развитие детей с синдромом Дауна в разных случаях сильно различается. На данный момент невозможно до рождения определить, как хорошо ребёнок будет обучаться и развиваться физически. Определение оптимальных методов проводится после рождения при помощи раннего вмешательства. Так как дети имеют широкий спектр возможностей, их успех в школе по стандартной программе обучения может сильно варьироваться. Проблемы в обучении, присутствующие у детей с синдромом Дауна, могут встречаться и у здоровых, поэтому родители могут попробовать использовать общую программу обучения, преподаваемую в школах.
В большинстве случаев у детей есть проблемы с речью. Между пониманием слова и его воспроизведением проходит некоторая задержка. Поэтому родителям рекомендуется водить ребёнка на обучение к логопеду. Мелкая моторика задерживается в развитии и значительно отстаёт от других двигательных качеств. Некоторые дети могут начать ходить уже в два года, а некоторые только на 4-м году после рождения. Обычно назначают физиотерапию, чтобы ускорить этот процесс.
Часто скорость развития речи и коммуникативных навыков задерживается и помогает выявить проблемы со слухом. Если они присутствуют, то при помощи раннего вмешательства это лечат либо назначают слуховые аппараты.
Детей с синдромом Дауна, учащихся в школе, обычно распределяют по классам по-особенному. Это обусловлено пониженной обучаемостью детей, страдающих синдромом Дауна, и очень вероятным отставанием их от сверстников. Требования в науках, искусстве, истории и других предметах могут быть для таких детей недостижимыми или достигнуты значительно позже обычного, по этой причине распределение положительно влияет на обучение, давая детям шанс. В некоторых европейских странах, как Германия и Дания, существует система «двух учителей», в которой второй учитель берёт на себя детей с коммуникационными проблемами и умственной отсталостью, однако это происходит в пределах одного класса, что препятствует увеличению умственного разрыва между детьми и помогает ребёнку развивать коммуникативные способности ещё и самостоятельно.
Как альтернатива методу «двух учителей» существует программа сотрудничества специальных и общеобразовательных школ. Суть этой программы заключается в том, что основные занятия для отстающих детей проводятся в отдельных классах, чтобы не отвлекать остальных учеников, а различные мероприятия, такие как: прогулки, занятия искусством, спортом, перемены и перерывы на питание проводятся совместно.
Дата добавления: 2022-11-11; просмотров: 92; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!