Диагностическая оценочная шкала Болезни Вильсона, Лейпциг, 2001
Болезнь Вильсона-Коновалова
(нарушения обмена меди)
Код по МКБ10: Е83.0
Этиология
Наследственное заболевание, аутосомно-рецессивный тип наследования.
ATP7B – ген экспрессируется, в основном, в печени и кодирует медьтранспортирующую АТФ-азу. Генетически детерминируемое снижение функции медь-транспортирующей АТФ-азы в результате молекулярных дефектов в гене АТР7В приводит к снижению гепатобилиарной экскреции меди и нарушению встраивания меди в церулоплазмин, в результате экскретируется и циркулирует апоцерулоплазмин (ненагруженный медью, срок полувыведения которого сокращается вдвое, что и объясняет гипоцерулоплазминемию), а медь накапливается в различных органах и тканях, преимущественно в печени, головном мозге, роговице глаза, почках, обеспечивая полиморфизм клинических появлений БВ.
Классификация:
Формы: бессимптомная, печеночная, неврологическая, смешанная
Классификация Коновалова 1960 г
| Брюшная (абдоминальная) форма | тяжелое поражение печени, приводящее к смерти до появления симптомов со стороны нервной системы; заболевают дети. Её продолжительность от нескольких месяцев до 3-5 лет при отсутствии терапии |
| Ригидно-аритмо-гиперкинетическая или ранняя форма | отличается быстрым течением; начинается также в детском возрасте. В клинической картине преобладают мышечная ригидность, приводящая к контрактурам, бедность и замедленность движений, хореоатетоидные или торсионные насильственные движения. Характерны дизартрия и дисфагия, судорожный смех и плач, аффективные расстройства и умеренное снижение интеллекта. Заболевание длится 2-3 года, без лечения заканчивается летально; |
| Дрожательно-ригидная форма | наиболее часто встречающаяся, начинается в юношеском возрасте, протекает медленнее, с периодическими ремиссиями и внезапными ухудшениями, сопровождающимися субфебрильной температурой. Характеризуется одновременным развитием тяжелой ригидности и дрожания. Дрожание очень ритмичное (2-8 дрожаний в секунду), резко усиливается при статическом напряжении мышц, движениях и волнении, в покое и во сне исчезает. Иногда обнаруживаются атетоидные хореоформные насильственные движения; наблюдаются также дисфагия и дизартрия. Средняя продолжительность жизни около шести лет |
| Дрожательная форма | начинается в возрасте 20-30 лет, протекает довольно медленно (10-15 лет и больше). Преобладает дрожание, ригидность появляется лишь в конце болезни. В ряде случаев наблюдается гипотония мышц; отмечается амимия, медленная монотонная речь, тяжелые изменения психики, часты аффективные вспышки. Наблюдаются эпилептиформные припадки. |
| Экстрапирамидно-корковая форма | встречается реже других форм. Типичные для гепато-церебральной дистрофии нарушения в дальнейшем осложняются апоплектиформно развивающимися пирамидными парезами, эпилептиформными припадками и тяжелым слабоумием (обнаруживаются обширные размягчения в коре больших полушарий). Длится 6-8 лет, заканчивается летально. |
Клинические признаки
|
|
|
|
|
|
| Поражение печени | - Бессимптомная гепатомегалия - Изолированная спленомегалия - Цитолитическая активность биохимических показателей - Стеатогепатит - Острый (фульминантный) гепатит - Острая печеночная недостаточность с гемолизом - Аутоиммуноподобный гепатит - Портальная гипертензия: ВРВП, спленомегалия, тромбоцитопения - Цирроз печени |
| Поражение ЦНС | - Двигательные нарушения (тремор, непроизвольные движения, гарушение походки) - Слюнотечение, дизартрия, дисфагия - Ригидная дистония - Псевдобульбарный синдром - Вегетососудистая дистония - Мигренеподобные головные боли - Бессонница - Дистонические атаки |
| Психиатрические симптомы | - Депрессия - Тревожные расстройства - Невротическое поведение - Изменения личности - Психоз |
| Другие системы | - Офтальмология: кольца Кайзера-Флейшера, «медная» катаракта - Гемолитическая анемия - Дерматологические проявления: «голубые ногтевые луночки» - Патология почек: аминоацидурия, нефролитиаз - Патология скелета: ранний остеопороз, артрит - Поражение сердца: кардиомиопатия, нарушения ритма - Панкреатит, желчнокаменная болезнь - Гипопаратиреоидизм, гигантизм - Нарушение менструального цикла, бесплодие, повторные выкидыши |
Кольца Кайзера-Флейшера – отложение меди на десцеметовой мембране роговицы (осмотр с помощью щелевой лампы).
|
|
|
Прогностический признак БВ:
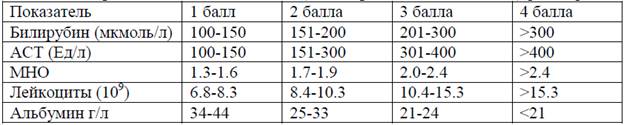
При 11 б и более высокая вероятность летального исхода без пересадки печени
Диагностическая оценочная шкала Болезни Вильсона, Лейпциг, 2001
| Признак | Баллы | ||
| 1.Кольца Кайзера-Флейшера - присутствуют - отсутствуют | 2 0 | ||
| 2.Нейропсихиатрическая симптоматика (изменения на МРТ) - выраженные - умеренные - отсутствует | 2 1 0 | ||
| 3.Церулоплазмин сыворотки крови - в норме >0,2 г/л (>20 мг/дл) - 0,1-0,2 г/л (10-20 мг/дл) - <0,1 г/л (<10 мг/дл) | 0 1 2 | ||
| 4.Кумбс-негативная гемолитическая анемия - присутствует - отсутствует | 1 0 | ||
| 5.Колич. определение меди в биоптатах печени (при условии отс. холестаза) - >250 мкг/г (4 мкмоль/г) - 50-250 мкг/г (0,8-4 мкмоль/г) - в норме (ниже 50 мкг/г) - Родамин – присутствуют позитивные гепатоциты | 2 1 -1 1 | ||
| 6.Экскреция меди с мочой (при отсутствии острого гепатита) - более 2 норм ИЛИ в норме, но увеличение более 5 норм при пробе с пеницилламином - 1-2 нормы - в норме | 2 1 0 | ||
| 7.Молекулярно-генетический анализ - гомозигота, компаунд-гетерозигота - гетерозигота - мутаций не обнаружено | 4 1 0 | ||
| Суммарные баллы:
4 и более: диагноз Болезнь Вильсона установлен; 3: диагноз Болезнь Вильсона вероятен, но требуется дальнейшее обследование пациента; 2 и менее: диагноз Болезнь Вильсона сомнителен. | |||
Обследование
1. ОАК (гемолитическая анемия, отрицательная проба Кумбса)
2. Б/х анализ крови: АЛТ, АСТ, билирубин общий, прямой, ЩФ, гамма-ГТ, церулоплазмин, медь сывороточная свободная, железо сыв, креатинин, мочевина, калий, натрий, общий белок, белковые фракции, СРБ, глюкоза, амилаза, холестерин. (увеличение печеночных/ почечных показателей, снижение церулоплазмина, медь норма или увелич)
3. ОАМ
4. Коагулограмма (печночная недостаточность, снижение ПТИ, фибриногена, увеличение ПВ, АЧТВ)
5. Группа крови, ВИЧ, вирусные гепатиты В, С, RW
6. суточная экскреция меди в моче (увеличение), медь разовой мочи (увеличение)
7. молекулярно-генетическое исследование
8. ЭКГ, РгОГК
9. ФЭГДС (ВРВП, портальная гастропатия)
10. УЗИ ОБП+почки (гепатомегалия, неоднородность печени, гиперэхогенности, спленомегалия, увеличение ВВ, СВ, асцит)
11. Фиброэластометрия печени
12. МРТ или КТ головного мозга (при неврологической симптоматике)
13. ЭЭГ (при неврологической симптоматике)
14. Консультация офтальмолога, осмотр роговицы с помощью щелевой лампы.
15. Консультация невролога
16. Чрескожная биопсия печени, (Биопсия печени под контролем ультразвукового исследования) с количественным определением содержания меди в ткани печени и гистологическим исследованием биоптата (Патолого-анатомическое исследование биопсийного (операционного) материала пункционной биопсии печени).
17. Консультация врача генетика (поиск среди родственников)
Лечение
Диетотерапия
- ограничиваются продукты с высоким содержанием меди (печень, креветки, орехи, шоколад, грибы).
- специальные фильтры для водопроводной воды
- Запрещается использовать медную посуду для приготовления и хранения пищи
- Необходимо избегать приема витаминных и минеральных препаратов, содержащих медь.
Хелатная терапия
- пеницилламин (купренил), принимается за 1 час до еды. Принимается пожизненно
· Начальная фаза: 250-500 мг/сут с постепенным (каждые 4-7 дней) увеличением дозы на 250 мг до лечебной дозировки 1000-1500 мг в сутки, которая дается в 2-4 приема.
· нормализация показателей обмена меди при двух последовательных исследованиях, выполненных с интервалом в 3 месяца – переход к поддерживающе теапии.
· Поддерживающая терапия 750-1000 мг/сут (На фоне поддерживающей хелатной терапии уровень экскретируемой суточной меди с мочой необходимо поддерживать в диапазоне 200-500 мкг (3-8 мкмоль) в сутки)
· При применении Пеницилламина развивается недостаточность пиридоксина, что требует назначения витамина В6 (пиридоксин) в дозе 25-50 мг/сут.
- Цинка сульфат (при непереносимости пеницилламина) 50 мг 3 раза в день за 30 мин до еды (При старте терапии уровень экскретируемой меди не должен превышать 100 мкг в сутки (1,6 мкмоль в сутки) [3], на поддерживающей терапии - в диапазоне 30-75 мкг в сутки (0,5-1,2 мкмоль в сутки); уровень меди в моче ниже 30 мкг в сутки указывает на передозировку препаратом)
- Триентин (триэтилен гидрохлорид) – при непереносимости пеницилламина 1-2 г в три приема за 60 минут до еды. В начальной фазе терапии триентин обычно назначается в дозе 750- 1500 мг/сут., в поддерживающей - в дозе 250-500 мг/сут., разделенной на 2-3 приема.
Беременность, лактация: снизить дозы хелаторов на протяжении всей беременности на 25-50% от исходной дозы. Терапию цинком можно сохранять без корректировки дозы. Следует проводить контроль лабораторных показателей обмена меди (не реже 1 раза в 3 месяца). Не рекомендуется совмещать прием пеницилламина с грудным вскармливанием, по поводу препаратов цинка недостаточно данных в настоящее время
Показания к пересадке печени
- развитие острой печеночной недостаточности
- неэффективность терапии хелаторами меди в течение нескольких месяцев у пациентов с декомпенсированным циррозом печени;
- возникновение тяжелой прогрессирующей печеночной недостаточности при самостоятельном прекращении лечения, прогрессирующих и необратимых неврологических нарушениях.
После трансплантации печени пациенты не нуждаются в хелатной терапии
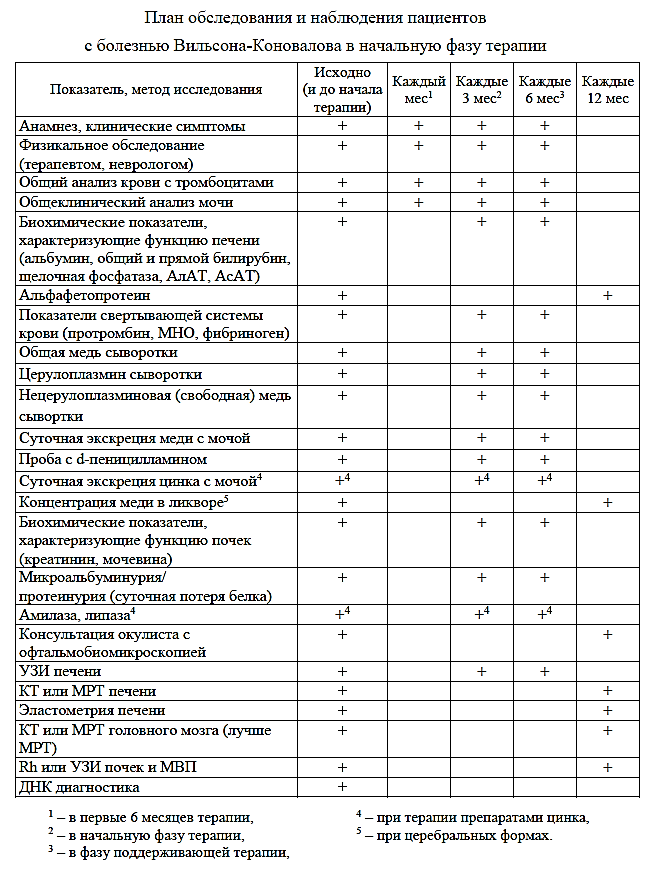
Наблюдение:
- Контроль лечения проводится 1 раз в неделю (ОАК, Б/х, коагулограма) на этапе старта терапии и в период повышения дозы пеницилламина, далее лабораторные и инструментальные исследования проводятся каждые 1-3 месяца до наступления ремиссии, и далее каждые 3-6 месяцев (возможно чаще по показаниям).
- офтальмолог, невролог 1 раз в год
- УЗИ ОБП, ФЭМ 1 раз в 6 мес
Дата добавления: 2021-12-10; просмотров: 11; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!