Клинико-цитогенетическая характеристика синдромов, связанных с аномалиями половых хромосом
Примеры хромосомных заболеваний.
Хромосомные болезни - это большая группа врожденных наследственных болезней. Хромосомные болезни занимают одно из ведущих мест в структуре наследственной патологии человека. По данным цитогенетических исследований среди новорожденных детей частота хромосомной патологии составляет 0,6-1,0%. Самая высокая частота хромосомной патологии (до 70%) зафиксирована в материале ранних спонтанных абортусов. Следовательно, большинство хромосомных аномалий у человека несовместимо даже с ранними этапами эмбриогенеза. Такие зародыши элиминируются во время имплантации (7-14-е дни развития), что клинически проявляется как задержка или выпадение менструального цикла. Некоторая часть эмбрионов гибнет вскоре после имплантации (ранние выкидыши). Сравнительно немногие варианты числовых аномалий хромосом совместимы с постнатальным развитием и ведут к хромосомным заболеваниям.
Хромосомные болезни появляются вследствие повреждений генома, возникающих при созревании гамет, в процессе оплодотворения или на ранних стадиях дробления зиготы. Все хромосомные болезни могут быть разделены на 3 большие группы:
1) связанные с нарушением плоидности;
2) обусловленные нарушением числа хромосом;
3) связанные сизменением структуры хромосом.
Аномалии хромосом, связанные с нарушением плоидности, представлены триплоидией и тетраплоидией, которые встречаются преимущественно в материале спонтанных абортусов. Отмечены лишь единичные случаи рождения детей-триплоидов с тяжелыми МВПР (множественными врожденными пороками развития), несовместимыми с нормальной жизнедеятельностью. Триплоидия может возникать как вследствие дигении (оплодотворение диплоидной яйцеклетки гаплоидным сперматозоидом), так и вследствие диандрии (обратный вариант) и диспермии (оплодотворение гаплоидной яйцеклетки двумя сперматозоидами).
|
|
|
Хромосомные болезни, связанные с нарушением числа отдельных хромосом в наборе, представлены либо целой моносомией (одной из двух гомологичных хромосом в норме) либо целой трисомией (тремя гомологами). Целая моносомия у живорожденных встречаются только по хромосоме X (синдром Шерешевского-Тернера), поскольку большинство моносомий по остальным хромосомам набора (Y хромосоме и аутосомам) погибают на очень ранних этапах внутриутробного развития и достаточно редко встречаются даже в материале спонтанно абортированных эмбрионов и плодов. Следует, однако, отметить, что и моносомия X с достаточно высокой частотой (около 20%) выявляется у спонтанных абортусов, что свидетельствует о ее высокой пренатальной летальности, составляющей свыше 99%. Целые трисомии встречаются по X, 8, 9, 13, 14, 18, 21 и 22 хромосомам. Наибольшая частота хромосомных нарушений – до 70% отмечается у ранних абортусов. Трисомии по 1, 5, 6, 11 и 19 хромосомам встречаются редко даже в абортивном материале, что свидетельствует о большой морфогенетической значимости этих хромосом. Более часто целые моно- и трисомии по ряду хромосом набора встречаются в мозаичном состоянии как у спонтанных абортусов, так и у детей с МВПР.
|
|
|
Хромосомные болезни, связанные с нарушением структуры хромосом, представляют большую группу синдромов частичных моно- или трисомии. Как правило, они возникают в результате структурных перестроек хромосом, имеющихся в половых клетках родителей, которые вследствие нарушения процессов рекомбинации в мейозе приводят к утрате или избытку фрагментов хромосом, вовлеченных в перестройку. Частичные моно- или трисомии известны практически по всем хромосомам, но лишь некоторые из них формируют четко диагностируемые клинические синдромы. Фенотипические проявления этих синдромов более полиморфны, чем синдромов целых моно- и трисомии. Отчасти это связано с тем, что размеры фрагментов хромосом и, следовательно, их генный состав, могут варьировать в каждом отдельном случае, а также тем, что при наличии хромосомной транслокации у одного из родителей частичная трисомия по одной хромосоме у ребенка может сочетаться с частичной моносомией по другой.
|
|
|
Синдром Патау (трисомия по хромосоме 13). Впервые описан в 1960 году. Популяционная частота 1 на 7800. Цитогенетические варианты могут быть различны: целая трисомия 13 (нерасхождение хромосом в мейозе, в 80% случаев у матери), транслокационный вариант (робертсоновские транслокации D/13 и G/13), мозаичные формы, дополнительная кольцевая хромосома 13, изохромосомы.
Для синдрома Патау характерны следующие диагностические признаки: микроцефалия, расщелина верхней губы и неба, низко посаженные деформированные ушные раковины, микрогения (недоразвитие нижней челюсти), полидактилия, флексорное положение пальцев рук, выпуклые ногти, поперечная ладонная складка, стопа-качалка. Из пороков внутренних органов отмечены врожденные пороки сердца (дефекты перегородок и крупных сосудов), незавершенный поворот кишечника, дивертикул Меккеля (локальное мешковидное выпячивание стенки подвздошной кишки), поликистоз почек, удвоение мочеточника. Наблюдается крипторхизм, гипоплазия наружных половых органов, удвоение матки и влагалища. Глубокая идиотия. Дети, в основном, умирают в возрасте до 1 года, чаще в первые 2-3 месяца жизни.
|
|
|
Синдром Эдвардса (трисомия по хромосоме 18). Описан в 1960 году. Популяционная частота составляет 1 на 6500. Цитогенетически в большинстве случаев представлен целой трисомией 18 (гаметическая мутация одного из родителей, чаще по материнской линии). Кроме того, встречаются и мозаичные формы, а транслокации наблюдаются очень редко. Критическим сегментом, ответственным за формирование основных признаков синдрома, является сегмент 18q11. Клинических различий между цитогенетическими формами не обнаружено.
Дети с синдромом Эдвардса имеют малую массу тела при рождении. Основными диагностическими признаками синдрома являются: долихоцефалия, гипертелоризм, низко посаженные аномальной формы уши, микрогнатия (недоразвитие верхней челюсти), микростомия, скошенный подбородок. Имеются аномалии развития конечностей: верхних - сгибательные деформации пальцев, перекрывание пальцев, сжатые пальцы рук, гипоплазия ногтей (особенно V пальца); нижних - короткий и широкий палец стопы, типичная форма стопы в виде качалки, кожная синдактилия стоп. Из внутренних пороков следует отметить комбинированные пороки сердечно-сосудистой системы, незавершенный поворот кишечника, пороки развития почек (чаще гидронефроз и подковообразная почка), крипторхизм. Отмечается задержка психомоторного развития, идиотия и имбецильность. Дети погибают, в основном, в возрасте до 1 года от осложнений, вызванных врожденными пороками развития.
Синдром Дауна (трисомия хромосомы 21).
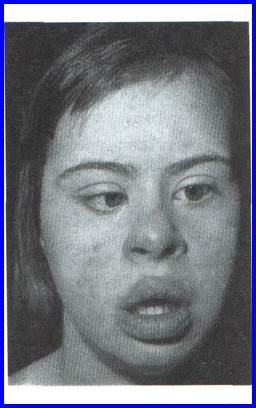
Впервые описан в 1866 году английским врачом Дауном. Наиболее часто встречающийся хромосомный синдром - популяционная частота составляет 1 случай на 600-700 новорожденных детей. Частота рождения детей с данным синдромом зависит от возраста матери и резко увеличивается после 35 лет. Цитогенетические варианты очень разнообразны, но около 95% случаев представлены простой трисомией 21 хромосомы, в результате нерасхождения хромосом в мейозе у родителей. Наличие полиморфных молекулярно-генетических маркеров позволяет определить конкретного родителя и стадию мейоза, в которой произошло нерасхождение (М1 – нерасхождение гомологичных хромосом 21 и М2 - нерасхождение хроматид). Этиологически важными факторами считаются внутри и внефолликулярное перезревание яйцеклетки, снижение числа или отсутствие хиазм в 1-м делении мейоза. Отмечены мозаичные формы синдрома (2%), робертсоновские транслокационные варианты (4%). Около 50% транслокационных форм наследуются от родителей и 50% являются мутациями de novo. Критическим сегментом, ответственным за формирование основных признаков синдрома, является область 21q22.
Основными диагностическими признаками синдрома являются: типичное плоское лицо, монголоидный разрез глаз, эпикант, открытый рот, макроглоссия и аномалии зубов, короткий нос и плоская переносица, избыток кожи на шее, короткие конечности, поперечная четырех-пальцевая ладонная складка (обезьянья борозда). Из пороков внутренних органов часто отмечаются врожденные пороки сердца и желудочно-кишечного тракта, которые и определяют продолжительность жизни больных. Умственная отсталость обычно средней степени тяжести. Дети с синдромом Дауна часто ласковые и привязчивые, послушные и внимательные.
Клинико-цитогенетическая характеристика синдромов, связанных с аномалиями половых хромосом
Синдром Шерешевского-Тернера (моносомия Х-хромосомы).
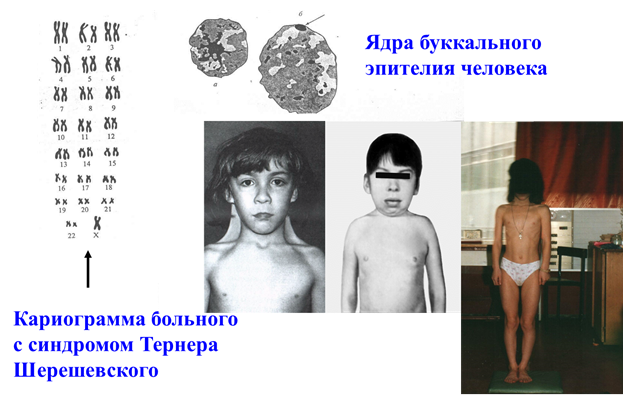
Это единственная форма моносомии у человека, которая может быть выявлена у живорожденных. Популяционная частота 1 на 3000 новорожденных. Кроме простой моносомии по X хромосоме, составляющей 50%, встречаются мозаичные формы, делеции длинного и короткого плеча X хромосомы, изо-Х-хромосомы, а также кольцевые X хромосомы. Интересно отметить, что мозаицизм 45, X / 46, XY составляет 2-5% от всех больных с этим синдромом и характеризуется широким диапазоном признаков: от типичного синдрома Шерешевского-Тернера до нормального мужского фенотипа.
Основными клиническими признаками заболевания являются: нанизм, крыловидные кожные складки на шее, короткая шея с низкой линией роста волос, отеки кистей и стоп новорожденных, бочкообразная грудная клетка, вальгусная девиация коленных и локтевых суставов. У больных выявляются первичная аменорея и половой инфантилизм, бесплодие, гиперпигментация кожи, снижение зрения и слуха. Часто встречаются врожденные пороки сердца и почек. Интеллектуальное развитие в пределах нормы.
Синдром полисомии Х-хромосомы. Популяционная частота 1 на 1000 новорожденных девочек. Цитогенетически выявляются формы 47,ХХХ, 48,ХХХХ и 49,ХХХХХ. С увеличением числа X хромосомы нарастает степень отклонений отнормы. У женщин с тетра- и пентасомией X описаны отклонения в умственном развитии, аномалии скелета и половых органов. Женщины с кариотипом 47,ХХХ в полной или мозаичной форме в основном имеют нормальное физическое и психическое развитие, а интеллект - в пределах нижней границы нормы. У этих женщин имеется нерегулярный менструальный цикл и вторичная аменорея, однако они могут иметь потомство.
Синдром Клайнфельтера. Описан в 1942 году. Популяционная частота 1 на 1000 мальчиков. Цитогенетические вариантысиндрома могут быть различны: 47,XXY; 48,XXYY; 48.XXXY; 49.XXXXY. Отмечены как полные, так и мозаичные формы.
Больные высокого роста с непропорционально длинными конечностями, выраженной гинекомастией и оволосением по женскому типу. В детстве отличаются хрупким телосложением, а после 40 лет страдают ожирением. Важными диагностическими признаками являются гипогонадизм и гипогенитализм. Характерно снижение полового влечения, импотенция и бесплодие. Коэффициент интеллекта ниже 80.
Синдром полисомии Y-хромосомы. Популяционная частота 1 на 1000 мальчиков. Цитогенетически отмечены полные и мозаичные формы. Большинство индивидов по физическому и умственному развитию не отличается от здоровых. Обычноони высокого роста и в 50% случаев могут иметь нормальное потомство. При данном синдроме имеются некоторые особенности поведения: склонность к агрессии, асоциальному поведению, гомосексуализму.
Дата добавления: 2021-04-24; просмотров: 100; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!