ПОЛУЧЕНИЕ ПОРОШКОВ ХИМИЧЕСКИМ ОСАЖДЕНИЕМ
Лекция 26.11.20
Никелевый порошокприменяется при очистке растворов для рафинирования никеля, в качестве катализатора при реакциях гидрогенизации и других, в производстве щелочных аккумуляторов (безламельные пластины), в магнитных устройствах, в качестве связующего при производстве твердых сплавов и т. д.
Электролитический метод получения никелевого порошка имеет наи~ большее применение потому, что порошок получается высокой чистоты, хорошо прессуется и спекается, имеет стандартные качества, позволяет использовать загрязненные исходные материалы.
Основными условиями для получения никелевого порошка электролизом являются: низкая концентрация ионов никеля в электролите, невысокая температура (30-40 °С.), высокая плотность тока и небольшая скорость циркуляции электролита.
В качестве электролита применяют разбавленные растворы сульфата никеля с добавкой аммонийных солей, препятствующих выпадению гидратов при малых катодных выходах по току (т. е. при выделении водорода) и связывающих никель в комплексные ионы.
Схематично процесс анодного растворения никеля можно представить следующей реакцией:
Ni ® Ni2+ +2e.
Анодный выход по току на никелевом растворимом аноде, особенно
в случае введения в электролит ионов хлора, обычно значительно превышает катодный выход по току. В этом случае в электролите увеличивается
концентрация ионов никеля, что может привести к получению компактного осадка на катоде. Во избежание этого принимаются особые меры для
поддержания постоянного состава электролита:
|
|
|
а) частичная пассивация никелевого анода (добавка NO3- ионов и др.), поддержание минимально допустимого количества ионов хлора в растворе;
б) охлаждение катода, изготовляемого в виде полого
кармана;
в) применение дополнительного, графитового анода,
кроме растворимого никелевого; подбором соотношения
действующих анодных площадей можно поддерживать
постоянную концентрацию ионов никеля в электролите, например, при соотношении площадей графитового к никеле5вому аноду равном 0,99 отношение выхода по току на катоде к выходу по току
на аноде равно 0,89; при соотношении площадей 2,75 отношение выходов
равно 1,06 и т. п.;
г) можно часть электролита периодически выводить на регенерацию с графитовым анодом, при этом можно получать на катоде обычный никелевый порошок, а на аноде – частично хлор.
Рекомендуемые условия электролиза: электролит — никель 3-6 г/л (в виде сульфата), (NН4)2SО4 –75-80 г/л, рН – 4,0-4,5; температура 30 °С; катодная плотность тока – от 2 (для более крупного) до 5 (для тонкодисперсного) тыс. А×м-2.
|
|
|
С увеличением температуры никелевый порошок, выделяющейся на катоде, становится более грубым. То же самое наблюдается при снижении плотности тока. На рис. 1 показано изменение среднего размера частиц никелевого порошка и катодного выхода по току от плотности тока.
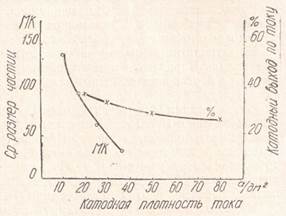
Рис.1. Влияние плотности ток на размер частиц
никелевого порошка и на выход по его току
После электролиза никелевый порошок промывают водой до отрицательной реакции на SO42-, затем сушат в автоклавах при 600-700 °С. Такой прием удобен в том отношении, что сначала водяные пары вытесняют воздух и предупреждают сильное окисление, затем начинает выделяться окклюдированный водород, который восстанавливает малейшие окислы. Никелевый порошок должен удовлетворять следующим условиям: влаги – не больше 0,2%, кислорода – не больше 0,1%, Ni + Со – 99,1%; С – не > 0,06%; Сu – не > 0,68%; Si – не > 0,05%; Fе – не > 0,2%;
S < 0,01%. Такой порошок можно хранить в сухом месте до 23 недель без окисления.
Активность никелевого порошка определяется по скорости выделения ям меди из раствора. Если через заданное время меди в растворе остается 5 мг/л, то порошки имеют активность ~ 85-90%, если 25 мг/л, то их активность равна – 50-55%.
Хранение никелевого порошка в растворе с рН меньше 3 позволяет сохранять активность в течение 6 месяцев.
|
|
|
Примеси кобальта, железа и марганца могут увеличивать активность никелевого порошка; примесь меди (в виде твердого раствора) снижает его активность.
Медный порошок широко применяется для изготовления пористых бронзовых вкладышей для подшипников, медно-графитовых щеток для электрических машин, сердечников для электротехнических устройств, всякого рода облицовок, поясков, токопроводящих частей и т. д.
Электролитический метод производства медных порошков дает высококачественный продукт и очень удобен, потому что легко может быть осуществлен в условиях обычного завода по электролитическому рафинированию меди.
Низкая концентрация ионов меди и значительная концентрация кислоты в сернокислом электролите, высокие плотности тока, невысокая температура, добавки сахара, глицерина и мочевины, применение переменного тока, вращающиеся и автоматически встряхиваемые катоды – вот далеко не полный перечень многих предложений.
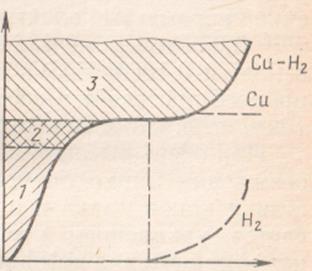
Было установлено, что между значениями плотностей тока, при которых происходит образование на катоде компактных осадков и порошков, лежит область плотностей тока, при которых уже не получаются гладкие компактные осадки, но и не появляются порошковые отложения. Эта область характеризуется увеличением поляризации с ростом плотности тока (рис. 2). Опыг показывает, что все факторы, способствующие снижению предельной плотности тока, будут благоприятствовать образованию более высокодисперсных порошковых осадков и, наоборот, условия, ведущие к повышению предельной плотности тока, спобствуют получению низкодисперсных, близких к компактным осадков.
|
|
|
| j к j пр j перех | Е Cu  E к
Рис.2. Параметры электролиза, характеризующие получения медного порошка. Зоны оразования компактных и рыхлых отложений: 1 – область образования компактных осадков; 2 – переходная E к
Рис.2. Параметры электролиза, характеризующие получения медного порошка. Зоны оразования компактных и рыхлых отложений: 1 – область образования компактных осадков; 2 – переходная  область;
3 – рыхлые порошкообразные отложения область;
3 – рыхлые порошкообразные отложения
|
Установлено, что понижение концентрации кислоты и температуры в сульфатном электролите ведет к увеличению зерен порошка, повышение плотности тока и уменьшение концентрации ионов меди – к уменьшению зерен. Увеличение скорости перемешивания ведет к
получению более однородных по величине, но более крупных зерен. Примеси ионов хлора, соединений фосфора, мышьяка, серы и кремния в электролите способствуют получению нитеобразных и ветвистых зерен малой прочности.
В качестве электролита применяют раствор: медь (в виде сульфата) – 10 г/л; серная кислота – 160 г/л, катодная плотность тока – около 2500 А×м-2; температура 50-58 °С, напряжение на ванне – около 1,5 В. Расход электроэнергии – около 1500 кВт×ч на1 тонну медного порошка.
Особое внимание уделяется стабилизации медного порошка, т. е. предохранению его от коррозии и окисления. Поверхность каждого зерна надо предохранять от влаги, в которой растворяются газы: О2, СО2, SО2, N2 и др., способствующие коррозии меди. Для этого рекомендуется создавать на зернах пленки высокомолекулярных, поверхностно-активных веществ. Таковые будут адсорбироваться своим полярным концом, другой же аполярный конец молекул будет придавать частицам диспергированного металла гидрофобные свойства. В качестве такой гидрофобизирующей добавки испытано и рекомендовано к применению хозяйственное мыло. Коррозионная стойкость медного порошка увеличивается от применения мыла в 50-70 раз. Требования, предъявляемые к медному порошку:
1. Зерна должны быть в форме дендритов, видимых при увеличении
в 100 раз; средний размер зерен должен быть в пределах 0,10-0,05 мм.
Остаток при просеве на сите в 325 меш должен составлять не более 35%,
на сите 200 меш – не больше 2% ,на сите 175 меш – 0,3%. Объемная
(насыпная) масса должен составлять 1,35-1,75 г×см-3.
2. Химический состав: Сu – не < 99,5%; далее не >: Fе – 0,022;
Рb – 0,05; Аs – 0,005; Sb – 0,01; SО42- – 0,01; Н2О – 0,05; остаток после
обработки НNО3 – 0,04%.
3. Цвет порошка должен быть бледно-розовым, окисляемость должна
быть очень малой.
Свинцовый порошок применяется в аккумуляторной промышленности, для производства тетраэтилсвинца, как добавка к маслам, идущим на смазку механизмов и т. д.
Предложено много способов для электролитического получения свинцового порошка. Например, из ацетатных растворов свинца с добавками перхлората или буры при 50-70 °С и катодной плотности тока 10-15 А×дм-2. В хлоридных электролитах получают черный губчатый свинец при малой плотности тока 0,2 А×дм-2.
Свинцовый порошок можно получить электролизом разбавленного раствора плюмбита натрия или калия при следующих условий электролиза: электролит: Рb (в виде Nа2РbО2) – 0,27 н; NаОН общ. – 2,54 н; Nа2СО3 – 0,55 н; температура 18-20 °C; плотность тока – 30-40 А×дм-2; продолжительность получения однородного, дисперсного осадка – 40-60 мин. Предлагается применение вращающегося, цилиндрического катода на горизонтальной оси.
Другие металлические порошки получаются в разных условиях электролиза: серебро – из разбавленных растворов нитрата; золото – из раствора хлорида золота при высокой плотности тока и низкой температуре; цинк – из раствора цинката, а также из слабокислых ванн в присутствии примесей; кадмий – из раствора сернокислого кадмия с добавками; олово – из щелочных и сернокислых растворов; сурьма – из растворов хлористой соли в виде так называемой «взрывчатой» сурьмы; кобальт - в условиях, аналогичных порошку никеля; хром – из раствора хлорида при высокой плотности тока и температуре, а также из ванн хромирования при огромных плотностях тока (до 30 000 А×м-2)в виде чешуек; теллур – из раствора двуокиси теллура в едком натре при 7-10 А×дм-2 и 25-30 °С.
Порошки бериллия, алюминия, марганца, хрома, циркония, титана, тория, ванадия, тантала, молибдена, вольфрама и урана можно получать электролизом их расплавленных солей и некоторых неводных растворов.
ПОЛУЧЕНИЕ ПОРОШКОВ ХИМИЧЕСКИМ ОСАЖДЕНИЕМ
МЕТАЛЛОВ ИЗ РАСТВОРОВ
Никелевый порошок
Патент США, № 3923496, 1975 г. Разработан способ получения никелевого порошка с размерами частиц в интервале 0,1—40 мкм, заключающийся в осуществлении реакции щавелевой кислоты с водным раствором азотнокислого никеля с целью получения оксалата никеля. Последний нагревают в атмосфере двуокиси углерода при температуре 450-850 °С в присутствии небольшого количества ингибитора роста зерен, такого как азотнокислый магний, с целью получения практически чистого никелевого порошка с близкими размерами частиц. Затем этот порошок нагревают в слабо окислительной атмосфере до умеренной температуры (<100 °С) для того, чтобы он стал непирофорным.
Пример 1. Получали никелевый порошок с размерами частиц 0,4 мкм. Для этого готовили раствор 35 кг специального химически чистого азотнокислого никеля Ni(NО3)2×6Н2О; его осветляли путем пропускания через небольшой фильтр-пресс, перекачивали в 285 л бак со стеклянной облицовкой и доводили до концентрации 2 М путем добавления воды и пара. После этого добавляли 0,035 % магнезии (отнесенной к никелю) в виде 4,1 г МgО, которая была полностью растворена в НNО3. Затем 16,7 кг технически чистой щавелевой кислоты (Н2С2О4×2Н2О) растворяли в горячей воде и доводили до концентрации 2 М в 76-л глиняном сосуде.
Раствор азотнокислого никеля нагревали до 97 °С, а раствор щавелевой кислоты – до 85 °С. Последний раствор равномерно добавляли к никелевому раствору в течение 10 мин; смесь поддерживали при температуре 97 °С в течение 20 мин. Осадок декантировали с тремя промежуточными промывками, а твердый оксалат никеля собирали в промывочном прессе; промывали и сушили при 110°С в течение ~ 18 ч.
Весь оксалат никеля (NiС2О4×2Н2О) в количестве ~20 кг поместили в большую вращающуюся обжиговую печь с внешним газовым подогревом. Затем порошок равномерно нагревали в атмосфере СО2 вплоть до достижения температуры 450 °С со скоростью ~100 °/ч. Обжиговая печь вращалась с частотой 2-3 об/мин и наклонялась назад и вперед, обеспечивая перемешивание, когда температура увеличивалась от 390 до 450 °С. После этого порошок охлаждали до температуры <1000С и обдували весь вечер слабым потоком воздуха с тем, чтобы он стал непирофорным. Наконец, его извлекали из обжиговой печи и пропускали через механический дезинтегратор.
Средние размеры кристаллов этого порошка (усредненные по объему) составляли 0,4 мкм, что было установлено методом измерения интенсивности окраски. Оценка размеров с помощью электронного микроскопа дала такой же результат. Анализ на содержание магния показал, что он присутствовал в количестве лишь 0,008 % (по отношению к никелю); это свидетельствует о том, что вместе с осадком оксалата никеля увлекалось лишь ~ 1/4 магния.
Пример 2. Когда предыдущий пример повторяли при температуре 500 °С, размер частиц порошка оказался равным 0,5 мкм, что было установлено методом измерения интенсивности окрашивания.
Пример 3. Получали никелевый порошок с размерами частиц 0,1 мкм. Растворяли в воде 70 кг специального химически чистого азотнокислого никеля Ni(NО3)2×6Н2О и отфильтровывали через осветляющий фильтр-пресс. Раствор перекачивали в бак из нержавеющей стали и доводили до концентрации 2 М путем добавления воды и пара. Затем добавляли 0,65 % магнезии (отнесенной к никелю) или 149,2 г МgО, которая была полностью растворена в НNO3.
В 150-л глиняном сосуде растворяли 33,3 кг технически чистой щавелевой кислоты (Н2С2О4×2Н2О) в горячей воде и доводили до концентрации 2М. Раствор азотнокислого никеля затем нагревали до 97 °С, а раствор щавелевой кислоты – до 85 °С. Щавелевую кислоту равномерно добавляли в течение 18 мин, и смесь выдерживали, в течение 20 мин при 97 °С. Промывку путем декантации повторяли трижды; оксалат никеля фильтровали через промывочный фильтр-пресс, промывали и сушили при 110°С в течение ~ 18 ч.
Оксалат никеля (NiС2О4×2Н2О) в количестве ~22,6 кг помещали затем в большую вращающуюся обжиговую печь, обогреваемую' газом. Порошок равноме рно нагревали со скоростью 100°/ч в атмосфере СО2 до тех пор, пока его температура не достигала 460 °С (печь вращалась с частотой 2-3 об/мин и покачивалась по достижении температурного интервала 390-460 °С для обеспечения перемешивания). Порошок охлаждали до температуры <100°С, затем обдували весь вечер слабым потоком воздуха с тем, чтобы он стал непирофорным, извлекали из печи и подвергали микроизмельчению.
Этот порошок имел средний размер кристаллов (усредненный по объему), равный 0,1 мкм, что было установлено с помощью электронного микроскопа. Анализ на содержание магния показал, что он присутствовал в количестве лишь 0,11 % (по отношению к никелю); это свидетельствует о том, что в осадок увлекалось всего лишь ~ 1/6 магния.
Размеры частиц дисперсных никелевых порошков очень быстро возрастали, если их нагревали выше ~400 °С. На рост размеров частиц влияют как время, так и температура; при этом максимальные размеры достигаются спустя 15-30 мин. Вследствие этого получение никелевых порошков с заданными размерами частиц оказывается технически неосуществимым, если не будут найдены ингибиторы роста частиц из группы, включающей магний, щелочные и щелочноземельные металлы. Если при нагреве присутствует один из представителей этой группы, даже в весьма незначительных количествах, то рост частиц заметно ограничивается.
Нагрев чистого оксалата никеля в неокислительной атмосфере при температуре ~450 °С сопровождается практически полным восстановлением оксалата никеля до никелевого порошка. Если ингибитор отсутствует, го размеры частиц продукта будут порядка 1 мкм, а если нагрев продолжать или увеличивать температуру, то размеры частиц будут довольно быстро увеличиваться. Однако в присутствии небольшого количества магния получают частицы намного меньших размеров. Взаимосвязь различных факторов, таких как процентное содержание ингибитора и темпеартура, более подробно иллюстрируется на рисунках, приводимых в патенте.
Медный порошок
Патент США, № 4047940, 1977 г. Описан процесс регенерации меди из растворов, содержащих медноаммиачные комплексы, путем восстановления меди в комплексах до одновалентного состояния с последующим ее осаждением в виде ацетиленида меди с помощью пропускания через раствор ацетилена. После отделения ацетиленида меди от раствора и тщательной промывки он реагирует с ацетонитрилом и кислотой с образованием ацетилена и медноацетонитрильного комплекса Сu(СН3СN)2+. Содержащий этот комплекс раствор затем подвергается однократному испарению с целью удаления ацетонитрила и превращения нестабильных ионов одновалентной меди в металлическую медь и двухвалентные ионы меди. Вслед за этим превращением компоненты разделяются и металлическая медь собирается.
Пример. Приблизительно 13,4 г основного карбоната двухвалентной меди (55,9 % Сu) и 10 г углекислого аммония растворяли вместе в 300 мл гидроокиси аммония (30 % NН3) и добавляли воду до общего' объема 1 л. Темно-голубой аммиачный раствор наливали в колбу для восстановления окисью углерода. Температуру в колбе поддерживали постоянной, равной 45±2 °С, а рН раствора корректировали до 10,5 путем регулирования относительных количеств подаваемых аммиака и окиси углерода. Находившийся в колбе воздух замещали окисью углерода, а отходящие газы пропускали через ротаметр.
Перемешивание начинали, когда воздух из колбы был полностью вытеснен. Для инициирования восстановления двухвалентной меди до одновалентной в колбу помещали кусок медной проволоки, предварительно очищенный путем погружения в раствор азотной кислоты. При поддержании слабого положительного потока окиси углерода через колбу отбирали пробы раствора с помощью герметичного шприца. Извлеченные пробы анализировали на содержание ионов одновалентной меди путем окисления стандартным раствором трехвалентного железа с последующим титрованием полученных ионов двухвалентного железа.
По завершении восстановления 600 мл бесцветного раствора одновалентной меди (содержавшего 4,7416 г Сu) перекачивали в колбу при сохранении слабого расхода аргона для предотвращения окисления. Перед введением раствора одновалентной меди в колбу воздух из нее вытесняли окисью углерода. В колбу с находившейся в ней порцией в 600 мл раствора одновалентной меди подавали предварительно очищенный ацетилен, и раствор подвергали умеренному перемешиванию. Реакция между ионами одновалетной меди и ацетиленом при данной температуре, т. е. 25-30 °С, протекала почти мгновенно.
Через несколько минут осаждение ацетиленида меди завершалось при слабом положительном потоке ацетилена и умеренном перемешивании. Пульпу затем фильтровали в атмосфере аргона, а фильтрат накапливали в сборной камере и анализировали методом атомной абсорбции с целью определения количества оставшейся в растворе меди. Отжатый осадок на фильтре промывали 200 мл разбавленного аммиачного раствора (рН = 9-9,5) и промывочный раствор анализировали методом атомной абсорбции с целью определения процентного содержания меди в нем.
Количество твердого вещества в отжатом осадке на фильтре определяли путем растворения взвешенного количества влажного ацетиленида в НС1 и анализа раствора на медь. Количество твердого вещества рассчитывали в предположении, что вся медь в нем грисутствует в виде Си2С2.
Затем полученные, как описано выше, 28,8 г отжатого на фильтре осадка переносили в четырехгорловую колбу с круглым дном. Работу с влажным ацетиленидом меди проводили так, чтобы был исключен или сведен к минимуму контакт с воздухом (для предотвращения окисления). К влажному ацетилениду в колбе добавляли раствор, содержавший 44,8 мл Н2О, 6 мл Н2SО4 (96,4 %) и 23 мл СН3СN и имевший температуру 40 °С. Перед этим для удаления всего кислорода подавали аргон с малым расходом.
Ацетиленид полностью растворялся за 25 мин при 40+2 °С и при этом получался светлый желтовато-прозрачный раствор. Ацетилен, выделявшийся при растворении ацетиленида в колбе, уносился гз нее потоком аргона; при желании его можно было улавливать в одной из двух систем газовой очистки.
По завершении растворения поток выходившего газа переключали из конденсатора-дефлегматора на спиральный конденсатор. Температуру водяной бани повышали с целью отделения путем дистилляции ацетонитрила от медного1 раствора, и при этом ионы одновалентной меди преобразовывались в металлическую медь и поны двухвалентной меди.
Азеотропная смесь СН3СN : Н2О=83,7 : 16,3 кипела при 76,5 °С. Нагрев колбы продолжали до тех пор, пока температура не достигала 89°С и при этой температуре собирали конденсат (32 мл). Остававшийся в колбе раствор (66 мл) содержал 2,051 г Сu2+. Металлическую медь собирали путем фильтрования, промывали водой, ацетоном и, наконец, бензолом, после чего сушили в атмосфере аргона.
Для определения общего количества полученной металлической меди пустую колбу промывали водой, а затем с помощью раствора
HNO3 растворяли любую медь, которая могла остаться на стенках. Концентрацию меди в этом растворе определяли методом атомной абсорбции, а общую массу регенерированной меди рассчитывали путем прибавления массы меди в этом растворе к массе собранного медного порошка. Общая масса металлической меди, полученной л таких условиях, составляла 1,899 г.
Дата добавления: 2020-12-22; просмотров: 362; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!