Аутосомно-рецессивные заболевания
Аутосомно-доминантные заболевания
При аутосомно-доминантном наследовании гетерозиготное носительство мутации оказывается достаточным для проявления заболевания. При этом мальчики и девочки болеют с одинаковой частотой. В количественном отношении доминантных заболеваний больше, чем рецессивных. В отличие от рецессивных доминантные мутации не приводят к полной инактивации функции кодируемого белка. Их эффект обусловлен либо снижением его количества (так называемая гаплонедоста-точность ), либо появлением у мутантного белка нового агрессивного свойства.
Вероятность рождения больных детей в браке гетерозиготного носителя доминантной мутации со здоровым супругом составляет 50%. Поэтому аутосомно-доминантные заболевания могут носить семейный характер и передаваться из поколения в поколение, причем среди родственников только со стороны одного из родителей больного. Такой тип передачи заболевания иногда называют наследование «по вертикали». Если оба родителя ребенка с доминантным заболеванием оказываются здоровыми, можно предположить, что болезнь развилась вследствие возникновения новой мутации в половых клетках одного из супругов.
По некоторым данным около 80% синдромов с аутосомно-доминантным типом наследования являются следствием мутаций de novo в половых клетках отцов. В этом случае риск повторного рождения больного ребенка такой же, как в любых других семьях. Исключением из этого правила являются доминантные заболевания с неполным проявлением или неполной пенетрантностью, когда на развитие заболевания дополнительно оказывают влияния какие-то внешние факторы или чаще состояния каких-то других генов. В этих случаях носители доминантной мутации могут быть здоровыми, а их дети больными или наоборот. Пенетрант-ность выше 60% является высокой степенью повторяемости заболевания в поколениях. Доминантный ген может обладать разной экспрессивностью, то есть внутри одной семьи картина заболевания может варьировать по степени тяжести и клиническим проявлениям. Напомним, что термины пенетрантность и экспрессивность в генетическую практику были введены известным отечественным генетиком Н. В.Тимофеевым-Ресовским. Об этом крупном ученом генетике и интереснейшем человеке петербургский писатель Даниил Гранин написал повесть «Зубр».
|
|
|
Доминантные мутации в гомозиготном состоянии у больных встречаются редко, и, как правило, они ассоциированы с более тяжелой клиникой. Так, при гетерозиготном носительстве доминантной мутации в гене рецептора липопротеинов низкой плотности у больных семейной гиперхолестеринемией ишемическая болезнь сердца и инфаркт миокарда развиваются в возрасте 30-40 лет, тогда как при гомозиготном носительстве - в первой декаде жизни. При доминантном типе наследования не происходит накопления мутаций в популяции, так как больные часто не оставляют потомства в силу тяжести своего состояния. Многие доминантные заболевания проявляются в достаточно позднем возрасте.В конце прошлого века было показано, что самыми распространёнными аутосомно-доминантными заболеваниями являются наследствен ные опухолевые синдромы. Их суммарная частота в различных популяциях достигает 1%, причем чаще всего обусловливающие их мутации передаются из поколения в поколение, а не возникают de novo.
|
|
|
Наследственные дисплазии соединительной ткани
Наследственные дисплазии соединительной ткани - это гетерогенная группа моногенных болезней, обусловленных присутствием мутаций в генах белков внеклеточного матрикса или ферментов их биосинтеза, а также в генах, участвующих в регуляции морфогенеза соединительной ткани. Большинство этих заболеваний наследуются по аутосомно-доминантному типу. Ведущая роль в поддержании структурной целостности различных соединительных тканей принадлежит коллагенам, большому семейству близкородственных внеклеточных матриксных белков, составляющему более 30% общей массы белков тела млекопитающих. Открытие около 40 коллагеновых генов и расшифровка их молекулярной природы создали предпосылки для изучения молекулярных основ этиологии и патогенеза наследственных коллагенопатий - гетерогенной группы из более чем 70 моногенных заболеваний.
|
|
|
Наиболее известным генетическим вариантом наследственной дисплазии соединительной ткани является синдром Марфана. Долгое время предполагали, что это заболевание обусловлено мутациями в одном из коллагеновых генов. Однако оказалось, что при синдроме Марфана первичным биохимическим дефектом является нарушение структуры фибриллина 1 - структурного белка микрофибриллярных эластических волокон внеклеточного матрикса. Наряду с этим, описаны другие аутосомно-доминантные заболевания, при которых у больных наблюдается марфаноподобный фенотип. Остановимся более подробно на этих двух группах наследственных дисплазии соединительной ткани.
Наследственные коллагенопатии
В настоящее время известно 27 различных типов коллагеновых белков. Каждый из них состоит из трех равномерно скрученных полипептидных альфа-цепей, образующих структуру, подобную трехгранному шнуру. Разные типы коллагенов могут быть образованы либо тремя одинаковыми альфа-цепями, либо двумя или тремя различными полипептидами в соотношении 2:1 или 1:1:1 соответственно. Каждая альфа-цепь кодируется собственным геном, поэтому разнообразие коллагеновых генов больше, чем разнообразие соответствующих белков. Биосинтез зрелых коллагенов сопровождается необычно большим числом пост трансляционных модификаций, так что на одной молекуле проколлагеновой полипептидной цепи осуществляется более 120 реакций. В этих превращениях принимают участие более десятка различных ферментов. Все зрелые коллагеновые белки способны к образованию крупных су-прамолекулярных агрегатов. На рис.45 показаны основные этапы биосинтеза коллагена.
|
|
|
Любая альфа-цепь содержит коллагеновой домен, на всем протяжении которого за исключением короткого С-терминального участка каждая третья аминокислота является глицином. Таким образом, молекулярная формула коллагенового домена может быть записана как (Gly-X-Y)n, где X и Y - аминокислоты не-Gly типа. Различные коллагеновые альфа-цепи различаются по количеству и протяженности (Gly-X-Y)-MOTHBOB в коллагеновом домене и по конкретному содержанию аминокислот в X и Y положениях. Присутствие глицина, самой маленькой из аминокислот, в каждом третьем положении коллагеновых полипептидных цепей существенно для их правильного скручивания в тройную спираль, так как глицин при этом занимает ограниченное пространство в центре триплекса. Поэтому любые мутации, приводящие к замене глицина на другую аминокислоту, будут сопровождаться локальными нарушениями структуры тройной спирали и дезорганизацией более крупных агрегатов коллагена. К тяжелым последствиям также приводят мутации, нарушающие структуру С-концевого участка адьфа-цепи, так как образование триплекса по типу «застежки-молнии» начинается именно с этого участка молекулы. Кроме того, именно в этой области локализованы сайты взаимодействия коллагена более чем с 50 другими белками. Патологический процесс оказывается менее тяжелым, если в результате мутации альфа-цепь полностью утрачивает способность участвовать в формировании зрелых коллагеновых молекул. Это мутации, сопровождающиеся преждевременной терминацией трансляции или затрагивающие N-концевые районы альфа-цепи коллагена. При этом в образовании триплексной структуры принимают участие только нормальные полипептиды, мутантные альфа-цепи в нее не входят и вскоре после синтеза подвергаются внутриклеточному протеолизу. В результате снижается скорость синтеза зрелых коллагеновых молекул, но их структура сохраняется нормальной, и они не утрачивают способность к образованию упорядоченных супрамолекулярных агрегатов. Доминантный характер заболеваний, обусловленных нарушением структуры коллагеновых молекул, объясняется тем, что присутствие, наряду с мутантными, нормальных альфа-цепей не предотвращает образования дефектов в фибриллах или других надмолекулярных комплексах коллагена. Заболевания, вызванные нарушением биосинтеза коллагеновых молекул и связанные с присутствием мутаций в генах соответствующих ферментов, наследуются по рецессивному типу.
Коллагены I, II и III типов являются мажорными и составляют более 90% всех коллагенновых белков. Они способны формировать крупные высоко организованные фибриллы, в которых отдельные молекулы коллагена располагаются четырехступенчатыми уступами. Остальные коллагеновые белки относятся к классу нефибриллярных коллагенов, формирующих мелкие фибриллы, либо листовидные мембранные образования.
Коллаген I типа экспрессируется повсеместно, но особенно обильно представлен в костной системе, сухожилиях и коже. Коллаген II типа является мажорным хрящевым коллагеном. Он также составляет основу стекловидного тела. Кроме того, в хрящевой ткани экспрессируются минорные коллагены IX, X, XI и XII типов. Эмбриональный мажорный коллаген III типа является основным компонентом стенок сосудов и кишечника. В базальных мембранах присутствует коллаген IV типа. V коллаген образует каркас внутри фибрилл мажорных коллагенов. Коллаген VI типа участвует во взаимодействии между фибриллами мажорных коллагенов и другими структурными компонентами внеклеточного матрикса. Коллагены VII и XVII типов присутствуют в эпидермальных кератиноцитах и являются компонентами кожных опорных фибрилл. Коллагены VIII и XVIII типов найдены в эндотелии сосудов и роговице, они участвуют в регуляции неоваскуляризации и образовании мембраны Десцемета. Остальные коллагены ассоциируются с мажорными коллагенами I и II типов, способствуя их взаимодействию с другими белками внеклеточного матрикса. Очевидно, что структурные дефекты коллагенов могут сопровождаться тяжелыми повреждениями соединительной ткани. В настоящее время мутации, ассоциированные с различными нозологическими формами наследственных коллагенопатий, найдены в 25 коллагеновых генах, участвующих в синтезе 13 различных типов коллагенов. Клинические проявления этих заболеваний хорошо коррелируют с характером экспрессии различных типов коллагенов и с исполняемыми ими функциями.
Синдром Элерса-Данло
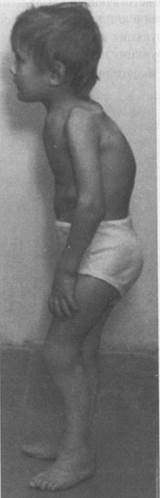
Классические варианты синдрома Элерса-Данло, характеризующиеся гиперрастяжимостью и истончением кожи, гипермобильностью суставов, неровным ростом зубов, деформацией ногтей, скелетными аномалиями, пролабированием клапанов сердца и др. клиническими проявлениями, обусловлены дефектами коллагена V типа.
|
|
|
|
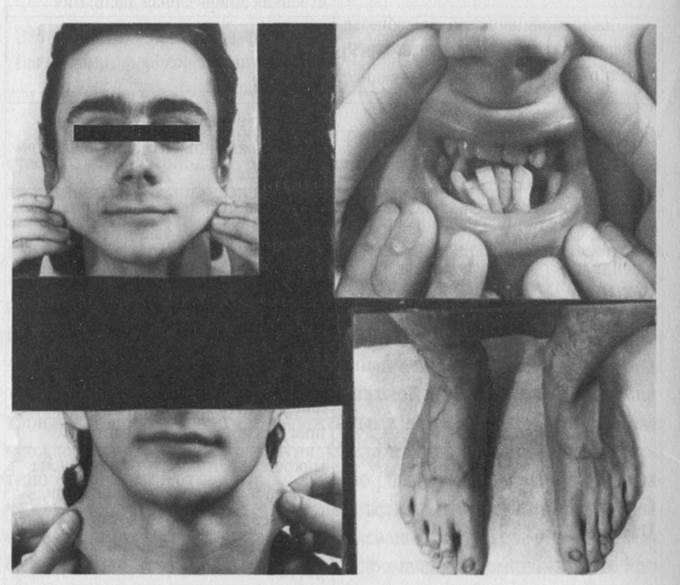
Рисунок 1. Больной с синдромом Элерса-Данло
Наиболее тяжелым является «артериальный» тип заболевания, так как может сопровождаться разрывами артерий и перфорацией внутренних органов. При этом дефектным оказывается коллаген III типа, обильно представленный в стенках сосудов и кишечника. При VII типе синдрома Элерса-Данло, характеризующимся сверх гиперрастяжимостью и лёгкой ранимостью кожи, выраженной гипермобильностью суставов, нанизмом и скелетными дисплазиями, найдены специфические мутации в генах COL 1 A 1 и COL 1 A 2 коллагена I типа. Все мутации, идентифицированные у больных с данным типом заболевания затрагивают сайт узнавания для одной из протеаз, участвующих в процессинга коллагена I, а именно в удалении N-концевого пропептида. Остальные варианты синдрома Элерса-Данло наследуются по аутосомно-рецессивному типу, так как большинство из них обусловлено мутациями в генах ферментов биосинтеза коллагена.
Сопутствующими симптомами многих вариантов наследственных коллагенопатий и в первую очередь синдрома Элерса-Данло, являются дистрофия ногтей, несовершенный дентиногенез, парадонтоз.
Пролапс митрального и других клапанов сердца также может сопровождать наследственные коллагенопатий. В частности этот симптом входит в структуру синдрома Стиклера и классических форм синдрома Элерса-Данло.
Синдром Марфана и другие наследственные синдромы с марфаноподобным фенотипом
Синдром Марфана впервые описан в 1896 году французским педиатром А. Б. Марфаном. У больных наблюдается одновременное поражение трех систем: опорно-двигательной, сердечно-сосудистой и органа зрения. Характерными клиническими проявлениями синдрома Марфана являются высокий рост, арахнодактилия (длинные, тонкие, «паукообразные» пальцы рук), гиперподвижность суставов, подвывих хрусталика и миопия, поражение крупных сосудов (аневризма аорты), порок сердца (пролапс митрального клапана. Каждый из этих симптомов может отличаться по степени тяжести и сочетаемости друг с другом у отдельных членов семьи.
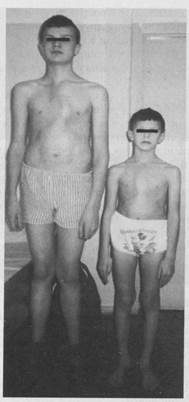
Рисунок 3. Родные братья с синдромом Марфана.
Для синдрома Марфана характерны выраженная плейотропия, варьирующая экспрессивность и высокая пенетрантность. Диагноз синдрома Марфана, ставится при наличии минимум пяти симптомов - аневризма аорты, вывих хрусталика, арахнодактилия, деформация грудины, кифосколиоз. При этом имеет место увеличение (в два раза и более) выведения с мочой глюкозоаминогликанов и их фракций. Особенно резко возрастает почечная экскреция хондроитин-4-6-сульфатов и в меньшей степени - гиалуроновой кислоты и гепаран-сульфата. В моче больных определяется также повышенное содержание (в два и более раз) аминокислоты оксипролина.
Распространенность заболевания в разных популяциях варьирует в пределах от 1:5 до 1:25 тысяч населения. Причиной развития классических форм заболевания 1 типа являются гетерозиготные мутации в гене фибриллина 1 - корового гликопротеина микрофибрилл эластических волокон внеклеточного матрикса, выполняющего в большинстве соединительных тканей архитектурные функции. Ген фибриллина 1 (FBN1) картирован в области 15q21.1, и в настоящее время в нем идентифицировано более 550 мутаций. Эти мутации обладают широким спектром клинических проявлений от изолированной эктопии хрусталика с мягкими скелетными проявлениями марфаноподобного типа до тяжелых неонатальных форм синдрома Марфана, заканчивающихся летальным исходом в течение первых двух лет жизни. При этом мутации, ассоциированные с тяжелыми формами синдрома, локализованы в определенных экзонах гена FBN1. Молекулярно-генетическая диагностика синдрома Марфана как в пренатальном, так и в постнатальном периоде принципиально возможна, но осложняется тем обстоятельством, что мутации в гене фибриллина уникальны, то есть описаны у больных только одной или значительно реже двух или нескольких неродственных семей. Подавляющее большинство из них приводят к классическим формам синдрома Марфана. Однако описаны другие аллельные варианты синдрома, выделяемые клиницистами в самостоятельные нозологические формы -табл. 1.
Важную роль в патогенезе синдрома Марфана играет трансформирующий фактор роста бета (Tgf|3), латентная форма которого непосредственно связана с фибриллином 1. Предполагается, что при снижении уровня фибриллина 1 повышается активность Tgf|3, следствием чего является высвобождение протеаз, участвующих в медленной деградации эластических волокон и других компонентов внеклеточного матрикса. Однако эти предположения требуют дополнительного экспериментального подтверждения.
Наряду с синдромом Марфана и его аллельными вариантами, известны другие марфаноподобные аутосомно-доминантные заболевания, не связанные с мутациями в гене FBN 1. Так, мутации в гене FBN 2, кодирующем изоформу 2 фибриллина, найдены у больных синдромом Билса - арахнодактилия, контрактуры суставов кистей рук, ки-фосколиоз, аномальная форма ушных раковин, генерализованная остео-пения, которая может способствовать развитию деформаций различных отделов опорно-двигательного аппарата. Для данного заболевания патология сердечно-сосудистой системы и органа зрения не характерны. При редком атипичном варианте синдрома Марфана найдены мутации в одном из генов коллагена I типа ( COL 1 A 2). Марфаноподобный фенотип наблюдается также при прогрессирующей диафизарной дисплазии 1 типа Камурати-Энгельманна, вызванной мутациями в гене Tgfpi -TGFBL Второй тип синдрома Марфана связан с мутациями в гене рецептора 2 Tgfpi - TGFBR 2. Мутации в гене TGFBR 1 найдены у больных синдромом Фурлонга - марфаноподобной болезнью II типа, сочетающейся с краниосиностозом и умственной отсталостью. Аллельным вариантом каждого из этих двух заболеваний является синдром аневризмы аорты Лоеса-Диетза. Этот синдром часто путают с синдромом Марфана, так как оба заболевания имеют перекрывающиеся спектры клинических проявлений. В таблице 1 представлены варианты наследственных скелетных дисплазии с марфаноподобным фенотипом.
Таблица 1.
Некоторые аллельные варианты синдрома Марфана и наследственные скелетные дисплазии с марфаноподобным фенотипом
| Нозологическая форма | Основные клинические критерии диагностики | Ген, первичный биохимический дефект |
| Синдром Марфана, тип 1 | дилатация или расслоение аорты, вывих/подвывих хрусталика, тяжёлая миопия, скелетные аномалии - высокий рост, долихостеномелия, арахнодактилия, деформация грудины, сколиоз, кифоз, дуральная эктазия и др. | FBN 1, фибриллин 1, коровый компонент микрофибрилл эластических волокон |
| Марфаноидный скелетный синдром | марфаноидный фенотип без сердечно-сосудистых и глазных аномалии | |
| Эктопия хрусталика, семейная | эктопия хрусталика, мягкие скелет- ные проявления, отсутствие кардио-васкулярной патологии | |
| MASS-синдром (Mitral valve, Aorta, Skeleton Skin) | пролапс митрального клапана, рас- ширение корня аорты, скелетные аномалии, истончение, атрофические полосы (стрии) кожи, ранняя миопия | |
| Синдром Марфана в сочетании с синдромом Шпринтцен-Голдберга | фенотипические проявления синдрома Марфана в сочетании с краниосиностозом, скафоцефалией, гипотонией, эндофтальмом, гипе- рэластичностью кожи, ректальным диастазом, вертикальной таранной костью и умственной отсталостью | |
| Синдром Вейла-Марчезани, аутосомно-доминантный | низкий рост, брахидактилия, туго- подвижность суставов и аномалии хрусталика |
Наследственные факоматозы
Факоматозы характеризуются сочетанным поражением нервной системы, кожных покровов и внутренних органов. Это название впервые предложил в 1923 году голландский офтальмолог J. van der Hoeve, описавший опухолевидные невоидные образования на сетчатке глаз при ту-берозном склерозе. Термин происходит от греческого слова "факон" - невус. Для многих факоматозов характерна варьирующая экспрессивность. Наряду с тяжелыми клиническими формами, отличающимися крайне неблагоприятным прогнозом, существуют стертые и олигосимптомные варианты. Наследуются факоматозы преимущественно по аутосомно-доминантному типу с неполной пенетрантностью, достигающей во многих случаях 75-90%. Факоматозы подразделяются на две большие группы: бластоматозы (нейрофиброматоз, туберозный склероз) и ангио-матозы (цереброретинальный ангиоматоз - синдром Гиппеля-Линдау, атаксия-телеангиэктазия, энцефалотригеминальный ангиоматоз и др.). Все факоматозы обусловлены мутациями в генах, относящихся к классу супрессоров опухолей.
Нейрофиброматоз I типа или болезнь Реклингхаузена-Уотсона -наиболее распространенная форма факоматоза с частотой среди населения 1:2500-3000. Клинически нейрофиброматоз I типа характеризуется тетрадой симптомов, описанных еще в 1930 F. J. Darier. (1) На коже туловища и конечностей больных наблюдаются разнокалиберные "кофейные" пятна, в динамике имеющих тенденцию к нарастанию по количеству и размеру. Кожные изменения могут также проявляться в виде участков депигментации, телеангиэктазий, гипертрихоза-рис.56. (2) Характерными клиническими проявлениями заболевания являются доброкачественные опухоли кожи и подкожной клетчатки - нейрофибромы, состоящие из смеси клеток Шванна и фибробластов. (3) Часто наблюдаются опухоли нервных стволов и окончаний, значительно варьирующие по форме, величине, количеству. (4) Для многих больных характерно отставание в физическом и умственном развитии различной степени выраженности.
Ген NF 1 характеризуется необычайно высокой частотой возникновения мутаций. Более 50% пациентов имеют вновь возникшие мутации, причем в подавляющем проценте спорадических случаев (90%) мутации имеют отцовское происхождение. Наиболее вероятным механизмом этой мутационной нестабильности, выражающейся в форме геномного импринтинга, является нарушения процесса метилирования гена NF 1. Белок, дефектный при нейрофиброматозе I типа получил название нейрофибромин. Он активно экспрессируется в эндотелии сосудов и в гладко-мышечных клетках. В 80% случаев мутации в гене NF 1 приводят к преждевременному окончанию синтеза белка. Большая часть из них представлена протяженными внутригенными перестройками. Соматические мутации в гене NF 1 идентифицированы в опухолевых клетках злокачественных меланом, нейробластом, анапластических астроцитом, спорадических карцином кишечника и других тканей.
Нейрофиброматоз II- типа встречается с частотой 1 на 33-40 тысяч новорожденных. Болезнь дебютирует, в среднем, в возрасте 21-22 лет. Выделяют центральную и спинальную формы заболевания. Клиническая картина определяется локализацией опухолей в веществе головного и спинного мозга. Опухоли мозга могут сопровождаться повышением внутричерепного давления, в результате которого появляется головная боль, рвота. Очаговая симптоматика зависит от места расположения опухоли и вовлечения в процесс черепно-мозговых нервов. Наиболее часто поражается слуховой нерв: невринома слухового нерва может быть одно- или двусторонней, причем в последнем случае может отмечаться грубая деформация продолговатого мозга. У больных с центральным нейрофиброматозом II типа, наряду с типичными двусторонними мультифокальными акустическими невриномами (опухолями восьмого краниального нерва, производными от клеток Шванна), могут развиваться менингиомы, билатеральные вестибулярные шваниомы, шваниомы дорзальных корешков спинного мозга и presenile lens opacities. Также как и при нейрофиброматозе I типа частыми являются изменения со стороны костной системы (задержка роста, сколиоз, кифоз, псевдоартроз, локальный гигантизм). У большинства пациентов с нейрофиброматозом II типа кофейные пятна и периферические нейрофибромы либо полностью отсутствуют, либо их число не превышает шести.
Наследование нейрофиброматоза II типа имеет те же особенности, что и при заболевании I типа, хотя и менее выраженные. Геномный им-принтинг выражается в более раннем дебюте и тяжелом течении заболевания при получении мутантного аллеля от матери, по сравнению с теми пациентами, которые унаследовали свою мутацию от отца. Потеря и/или аберрантное состояние локуса NF 2 является важным элементом развития не связанных с нейрофиброматозом II спорадических швани-ом и менингиом, которые вместе составляют около 30% всех первичных опухолей мозга. Так что продукт гена NF 2 также относится к супрессорам опухолей.
Туберозный склероз, описанный французским неврологом D.-M. Bourneville (1880) и английским дерматологом J. J. Pringle (1890), представляет собой гетерогенную группу аутосомно-доминантных заболеваний с неполной пенетрантностью и варьирующей экспрессивностью. Популяционная частота заболевания различна в разных возрастных группах - 1 на 30 000 среди взрослых и вдвое больше - 1 на 15 000 - среди детей в возрасте до 5 лет.
Туберозный склероз или эпилойя (эпилепсия плюс анойя - умственная отсталость) характеризуется триадой симптомов: а) кожными изменениями б) судорожными припадками; в) психическими расстройствами со значительным снижением интеллекта. Клинические проявления заболевания складываются в зависимости от преимущественного поражения головного мозга (в виде разрастания глии и появления атипичных муль-типолярных клеток в области бугорков), либо кожных покровов. Для последних характерно сочетание пигментированных пятен с участками депигментации в различных отделах туловища и конечностей. Специфичны изменения кожи, часто наблюдаемые в поясничной области в виде «шагреневой кожи». Возможно проявление фиброзного ангиоматоза в области крыльев носа и подбородка. Типичны ахромичные листовидные пятна, околоногтевые фибромы, аденоматозные разрастания сальных желез в виде «adenoma sebaceum» на спинке носа и в виде «бабочки» на щеках. У большинства больных уже в детском возрасте имеется та или иная степень снижения интеллекта, нарастающая в процессе жизни больного. Умственная отсталость встречается примерно у 70% больных и усугубляется вследствие деструкции мозга. Возможны изменения со стороны глаз в виде застойных сосков или атрофии зрительных нервов, эндокринные нарушения, пороки развития внутренних органов.
Первыми признаками заболевания у детей в возрасте 3-4-х месяцев могут явиться судорожные припадки тонического характера, затем они становятся полиморфными, плохо поддаются лечению. Иногда на глазном дне в области диска зрительного нерва обнаруживаются специфические разрастания, носящие название «тутовая ягода». Для младенческой формы заболевания характерны кардиальные и глазные гамартомы. Неврологические и психические нарушения являются результатом гамар-томных туберозных образований по ходу мозговых оболочек, извилин коры головного мозга. Чаще они появляются в области базальных ганглиев, стенок желудочков мозга, реже в области мозжечка, продолговатого мозга. Гамартомы представляют собой скопления атипичных ги гантских ганглиозных клеток, а в стенках желудочков содержат, кроме того, ангиоматозную ткань. Возможно вовлечение в опухолевый процесс других органов - почек, печени, сердца с последующей склонностью к малигнизации.
Туберозный склероз представляет собой генетически гетерогенную группу аутосомно-доминантных заболеваний со сходной клинической картиной. Примерно 40% семей с туберозным склерозом имеют I тип заболевания, обусловленный мутациями в гене TSC 1, локализованном в области 9q32-34. В подавляющем большинстве оставшихся семей болезнь обусловлена мутациями в гене TSC 2, расположенным в области 16р13.3. Это II тип туберозного склероза. Белки, кодируемые генами TSC 1 и TSC 2, были названы гамартином и туберином соответственно. Эти два белка взаимодействуют друг с другом с образованием туберин-гамартинового комплекса, участвующего в негативной регуляции инсу-линового сигнального пути. Гиперэкспрессия в системе in vitro каждого из генов TSC подавляет рост и пролиферацию клеток, а также изменяет их морфологию.
В гене TSC 2 обнаружено несколько сотен различных мутаций, значительная часть которых составляют протяженные внутригенные деле-ции. Подавляющее большинство мутаций гене TSC 1 приводит к образованию укороченных форм белка - это небольшие делеции, инсерции и нонсенс мутации. Для туберозного склероза характерна высокая частота возникновения новых мутаций, достигающая по разным оценкам 60-65%. В целом, мутации в генах TSC 1 и TSC 2 объясняют подавляющее большинство семейных и примерно половину спорадических вариантов туберозного склероза с превалирующим вкладом гена TSC2 во втором случае. Клиническая диагностика двух генетических форм заболевания, обусловленных мутациями в генах TSC 1 и TSC 2, практически невозможна, хотя очевидно, что умственная отсталость значительно чаще встречается у носителей мутаций в гене TSC 2. Молекулярная диагностика туберозного склероза достаточно трудоемка из-за генетической гетерогенности заболевания, большого количества экзонов и разнообразия мутаций в каждом из двух генов.
Аутосомно-рецессивные заболевания
Аутосомно-рецессивные заболевания проявляются только при гомозиготном носительстве мутантных аллелей. При этом происходит частичная или полная инактивация функции мутантного гена. Одну из мутаций больной ребенок наследует от матери, другую - от отца. Родители больного, будучи сами здоровы, являются гетерозиготными носителми мутации. Вероятность рождения больного ребенка в такой семье в соответствии с законом Менделя составляет 25%. Девочки и мальчики болеют с одинаковой частотой, при этом рождение больного ребенка совершенно не зависит от возраста родителей, очередности беременности и родов. Часто в одной семье может наблюдаться несколько больных сибсов. Две трети здоровых детей в браке гетерозиготных родителей также оказываются гетерозиготными носителями мутации. В браке гетерозиготного носителя рецессивной мутации с супругом, не имеющим мутантного аллеля, все дети будут здоровы, но половина из них окажутся гетерозиготными носителями мутации. Анализ родословных больных с аутосомно-рецессивными заболеваниями показывает, что часто (примерно в 60%) родители таких больных являются родственниками или их предки происходят родом из одного села или района, что так же является косвенным признаком инбридинга.
Муковисцидоз
Самым распространенным аутосомно-рецессивным заболеванием детского возраста среди представителей белой расы является муковисцидоз или кистозный фиброз поджелудочный железы. Впервые заболевание описано в 1938 году американским паталогоанатомом Д. Андерсон. Согласно зарубежной статистике частота муковисцидоза у жителей Западной Европы, в среднем, составляет 1 на 2-3 тысячи новорожденных, в России она меньше - 1 на 3-5 тысяч. Наблюдаются значительные географические и этнические различия по частоте муковисцидоза. В странах Африки и Азии муковисцидоз почти не встречается, тогда как каждый 20-й (5%) житель Западной Европы является гетерозиготным носителем мутации в гене муковисцидоза ( CFTR ). Такую высокую распространенность мутантных аллелей в гене CFTR связывают как с «эффектом основателя», так и селективным преимуществом гетерозигот, обусловленным устойчивостью к холере, туберкулезу, токсическим формам гриппа. С другой стороны, у гетерозигот вдвое выше частота хронического панкреатита.
Основной патогенетический механизм заболевания - увеличение вязкости секрета, выделяемого слизеобразующими железами бронхов, кишечника, поджелудочной железы, семявыводящих канальцев, сопровождающееся закрытием многих протоков в этих органах. В частности, нарушается естественный процесс очищения бронхов, что приводит к их воспалению. Воспаление сопровождается отеком легких и увеличением продукции аномально вязкого секрета. Повышенная вязкость секрета в желудочно-кишечном тракте сопровождается изменением водно-электролитного компонента панкреатического сока, его сгущением и затруднением выделения в просвет кишечника. В результате нарушается формирование каловых масс с последующей кишечной непроходимостью и происходит фиброзно-кистозное изменение ткани поджелудочной железы.
Минимальными диагностическими симптомами муковисцидоза являются рецидивирующие легочные, чаще всего синегнойные инфекции, нарушение функции кишечника и поджелудочной железы, отставание в физическом развитии. Характерными признаками заболевания считаются большое количество неперевариваемого жира в копрограмме больного и повышение концентрации ионов натрия и хлора при проведении потовой пробы. У некоторых больных уже при рождении наблюдается кишечная непроходимость, обусловленная мекониальным илеусом. Такие больные требуют срочного оперативного вмешательства. Иногда мекониальный илеус у плода с муковисцидозом можно обнаружить при ультразвуковом исследовании уже во 2-3-м триместрах беременности. Выделяют три клинические формы муковисцидоза: легочную (15-20% случаев), кишечную (10%) и смешанную. Описаны также стертые формы заболевания, выявляющиеся у взрослых.
Обследованию на наличие муковисцидоза подлежат следующие группы лиц: (1) больные с рецедивирующими бронхолегочными заболеваниями, синегнойной инфекцией, астмой, аллергозами; (2) лица с заболеваниями желудочно-кишечного тракта (склонные к запорам, колитам, хроническому панкреатиту, рецидивирующей кишечной непроходимости; новорожденные с мекониальным илеусом, перитонитом; дети с большим животом и низкой массой тела (при нормальном аппетите);больные циррозом печени неясного генеза; (3) мужчины с бесплодием после исключения других причин (у 98% мужчин больных муковисцидозом имеется сужение или атрезия семявыводящего протока). Муковисцидоз входит в программу неонатального скрининга новорожденных.
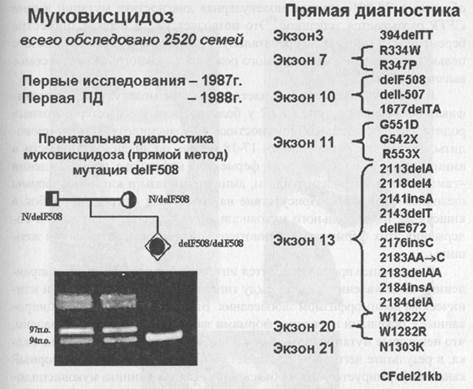
Ген муковисцидоза был картирован в 1985 году в длинном плече хромосомы 7 в области 7q31.2. В 1989 году он был идентифицирован, и это открытие сопровождалось одновременной публикацией в одном из самых престижных журналов мира («Science» - Наука) трех статей. Первая статья была посвящена самому гену муковисцидоза. Группе канадских исследователей под руководством Л. Ч. Тсуи удалось выделить и определить нуклеотидную последовательность кДНК гена муковисцидоза ( CFTR ). Были определены экзон-интронные границы и регуляторные области гена. Вторая статья была посвящена белку, кодируемому геном CFTR и являющимся первичным биохимическим дефектом у больных муковисцидозом. Оказалось, что этот трансмембранной белок, локализованный на апикальных мембранах экзокринных желез эпителия, участвует в регуляции проводимости ионов хлора и сам выполняет функции хлорного канала. В третьей статье был проведен анализ мутаций в гене CFTR . Как только становится известна нуклеотидная последовательность кодирующей области гена, сразу же можно задаться вопросом, а что случилось у больного, чем ген больного человека отличается от нормального гена? Используя нормальную кДНК гена CFTR в качестве ДНК-зонда, удалось выделить мутантную кДНК из эпителия бронхов девочки, больной муковисцидозом. На следующем этапе была определена полная нуклеотидная последовательность этой кДНК. Оказалось, что больная девочка гомозиготна по специфической мутации в гене CFTR -делеции трех нуклеотидов в 10-м экзоне, сопровождающейся отсутствием фенилаланина в 508 положении соответствующего белка - delF508. Частота этой мутации у больных муковисцидозом в Канаде, Северной Америке и Северной Европе достигает 80%. Мутация delF508 легко диагностируется методом ПЦР и электрофоретического разделения ампли-фицированных фрагментов ДНК. На рис.57 представлены мутации в гене CFTR , диагностируемые в лаборатории пренатальной диагностики наследственных и врожденных заболеваний ИАГ им. Д. О. Отта РАМН, возглавляемой чл-корр. РАМН проф. В. С. Барановым, указано количество обследованных семей больных муковисцидозом и дан пример пренатальной диагностики delF508. Рисунок 57. Диагностика мутаций в гене CFTR у больных муковис-цидозом (данные 2002 г.).
В настоящее время у больных муковисцидозом идентифицировано более 1000 разных мутаций в гене CFTR , главным образом, миссенс-типа. Однако самой распространенной остается delF508. Ее частота у больных муковисцидозом в разных популяциях варьирует от 30% до 80%. В Европе наблюдается определенный градиент распространения этой мутации с севера на юг: в Дании ее частота достигает 85%, в Италии снижается до 50% и в Турции - до 20-30%. В славянских популяциях частота delF508 среди больных муковисцидозом составляет около 50%. К мажорным мутациям относятся миссенс-мутации W1272X (встречается более чем в 30% случаев у больных муковисцидозом, принадлежащих этнической группе евреев ашкенази), G542X, G551D, R117H, R334W и др. Таким образом, в 70-80% случаев молекулярная диагностика мутаций в гене CFTR оказывается успешной. Это позволяет уже в первом триместре беременности проводить пренатальную диагностику муковисцидоза с целью предупреждения повторного рождения больного ребенка в семье высокого риска.
В тех случаях, когда не удается провести молекулярную идентификацию мутаций в гене CFTR у больного или у его гетерозиготных родителей, пренатальная диагностика муковисцидоза может проводиться при сроке беременности 17-18 недель по анализу активности в амниотической жидкости ряда ферментов кишечного происхождения -гамма-глютамилтранспептидазы, аминопептидазы и кишечной формы щелочной фосфатазы. Присутствие на этом сроке слизистых пробок в кишечнике плода больного муковисцидозом приводит к снижению содержания этих ферментов в амниотической жидкости беременной женщины.
В настоящее время проводятся интенсивные исследования, направленные на выявление связей между типами мутаций в гене CFTR и клиническим полиморфизмом заболевания. Выявлены мутации, ассоциированные с тяжелыми и мягкими формами заболевания. Было установлено, что некоторые мутации, в том числе delF508, нарушают процессинг белка, в результате чего он не достигает апикальной мембраны и хлорный канал не формируется. Этим объясняется тяжелая клиника муковисцидоза при подобных нарушениях. Другие мутации (R117H, R334W, R347P), выявленные при более мягких формах муковисцидоза, не затрагивают процессинг белка, хлорный канал формируется, но работает менее интенсивно. Так, например миссенс-мутация Rl 17Н обнаружена у мужчин, страдающих бесплодием в силу закупорки семявыводящих канальцев. При этом клиника муковисцидоза у таких пациентов, как правило, отсутствует или очень стерта. То есть, у носителей мутации R117H вязкость аномального секрета, выделяемого экзокринными железами эпителия, повышена настолько незначительно, что это не приводит к аномальным процессам в легких, поджелудочной железе или в кишечнике, но это повышение достаточно для формирования непроходимости семявыводящих канальцев (vas deferens).
С использованием техники трансгеноза в различных лабораториях США и Великобритании были сконструированы модельные линии мышей с мутациями в гене муковисцидоза, в том числе и такими, которые были идентифицированы у больных. Показано, что различные мутации по-разному влияют на фенотип животных. У мышей некоторых трансгенных линий отмечено преимущественное поражение легких, тогда как в других линиях - поджелудочной железы и кишечника. В одной линии наблюдали гибель большого числа зародышей от причин, сходных с ме-кониальным илеусом. Таким образом, эти линии представляют собой идеальные модели не только для изучения молекулярных основ патогенеза муковисцидоза, но и испытания различных программ терапии этого тяжелого заболевания.
В настоящее время разработаны эффективные методы этиопато-генетического лечения, позволяющие значительно увеличить продолжительность жизни больных муковисцидозом. Лечение больных муко-висцидозом показано проводить в специализированных региональных центрах. Общая схема лечения включает применение муколитических средств, антимикробных препаратов, ферментов поджелудочной железы, витаминов, в сочетании с применением кинезо- и физиотерапии. Ведение больных муковисцидозом проводится согласно национальным рекомендациям.
Дата добавления: 2019-11-25; просмотров: 200; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!