Определение образования, свойств, методов регулирования и исследования лиофильных и лиофобных дисперсных систем. Область и условия их применения.
Образование частиц дисперсной фазы приводит, как отмечалось в гл. I, к увеличению свободной энергии системы за счет возрастания свободной поверхности энергии ДУ, = оА5 при увеличении площади межфазной поверхности S. Вместе с тем, образование частиц дисперсной фазы, включающихся в самостоятельное броуновское движение, вызывает рост энтропии системы Д$ и уменьшение свободной энергии системы на величину 7Д?, что, как будет видно из дальнейшего изложения, может существенно изменять соотношение между энергиями макрофаз и дисперсной системы.
Впервые роль теплового движения частиц в термодинамике образования коллоидных систем была рассмотрена М. Фольмером (1927 — 1931). К сожалению, примерно в то же время А. Марх, сопоставив энтропию коллоидной системы с поверхностной энергией частиц дисперсной фазы, пришел к ошибочному выводу, что роль энтропийных эффектов пренебрежимо мала; это существенно замедлило развитие теории дисперсных систем. Ошибочность вывода Марха была связана с тем, что он ограничился анализом систем с достаточно высокой межфазной энергией (выше 1 мДж/м2, см. ниже).
Представления Фольмера были развиты в работах Ребиндера и Щукина[1], где дано общее и последовательное рассмотрение роли энтропийного фактора в термодинамике дисперсных систем, которое определило пути дальнейших исследований в этом направлении.
Следуя логической схеме этих авторов, оценим прирост энтропии при образовании дисперсной системы при диспергировании стабильной макрофазы. Для этого будем рассматривать дисперсную систему, содержащую o/Vi частиц (или А| = oV|/NA молей частиц) в Ы2 молях растворителя, как идеальный или регулярный раствор. Тогда увеличение энтропии при образовании дисперсной системы можно выразить как увеличение энтропии при смешении;
|
|
|
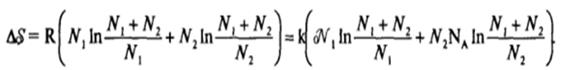
В реальной коллоидной системе число частиц дисперсной фазы много меньше, чем число молекул растворителя, т. е. N/N2 << 1. Так, при 0,1 %-ном объемном содержании частиц дисперсной фазы (с радиусом г и 1(Г8 м) отношение N/Ni составляет примерно 1(Г8. Учитывая, что ln(l + N/Nj) « Аг|/Л,2, выражение для AS в этом случае можно записать в виде:

где
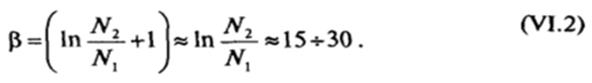
Общее изменение свободной энергии при образовании дисперсной системы с г можно представить в виде
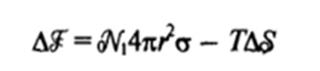
или приближенно, включая случай несферических частиц с линейным размером d,
где а — коэффициент формы.
Чтобы описать возможность образования термодинамически равновесной коллоидной системы, рассмотрим, как зависит от размера частиц или, что более наглядно, от логарифма размера (lg d), величина АЖ — разность свободных энергий дисперсной системы определенного состава, и макрогетерогенной системы того же состава (рис. VT-1).
|
|
|
Если поверхностная энергия велика, то по мере уменьшения размера частиц происходит рост энергии системы, и роль энтропийного фактора при всех размерах частиц, заметно превышающих молекулярный, пренебрежимо мала (рис. VI-1, кривая I). Для получения такой лиофобной дисперсной системы из соответствующих макроскопических фаз нужно затратить большую работу, например в виде работы механического диспергирования.
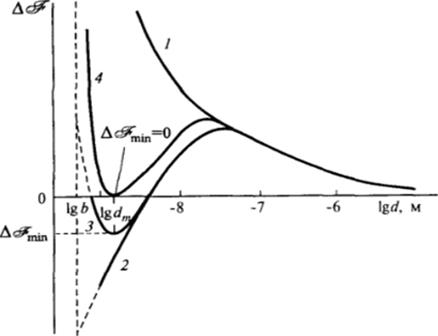
Рис. VI-1. Зависимость изменения свободной энергии ЬЗ монодисперсной системы от логарифма диаметра частиц d
При малой поверхностной энергии поверхности раздела фаз существенным становится учет прироста энтропии системы. При этом выражение (VI.3) принимает вид:
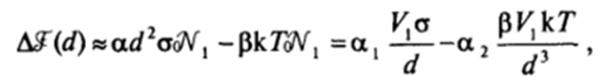
где численные коэффициенты а, и определяются формой частиц. В соответствии с этим выражением при малых размерах частиц должно преобладать второе, энтропийное слагаемое, и может появиться область состояний, для которых < 0, т. е. термодинамически более выгодных (имеющих более низкую свободную энергию), чем исходные макроскопические фазы.
В простейшем случае, когда размер частиц не влияет на величину a (o(d) = const), кривая AJ(1 gd) имеет только максимум (рис. VI-1, кривая 2), так что образование молекулярного раствора (d = Ь) термодинамически выгоднее, чем возникновение коллоидной системы. Чтобы более выгодным было образование коллоидной системы, а не молекулярного раствора, на кривой A?(lgt/) должен быть минимум в области dm, заметно превышающей размер молекул b (рис. VI-1, кривая 3). Возникновение такого минимума может быть связано с резким ростом величины а по мере приближения размеров частиц к молекулярным; возможная причина такого роста будет рассмотрена в следующем параграфе. Этот минимум отвечает наиболее термодинамически выгодному состоянию системы.
|
|
|
Если уменьшение свободной энергии системы из-за прироста энтропии компенсирует ее увеличение, связанное с развитием новой поверхности, так что в минимуме кривой №(gd) изменение свободной энергии оказывается отрицательным Akinin < 0, то образование коллоидной системы оказывается термодинамически выгодным процессом и может происходить самопроизвольно. При постоянном составе фаз условие термодинамической выгодности образования дисперсной системы из макрофазы имеет вид:
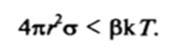
По Ребиндеру и Щукину, это условие имеет простой физический смысл: если межфазное натяжение мало, то возможно самопроизвольное отщепление частиц коллоидных размеров от макрофазы, поскольку работа, затрачиваемая на образование новой поверхности, компенсируется выигрышем энергии в результате прироста энтропии из-за участия образующихся частиц в тепловом движении. В соответствии с (VI.3) такой процесс самопроизвольного диспергирования макрофазы оказывается выгодным, если поверхностное натяжение не превышает критического значения ас:
|
|
|
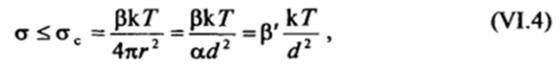
где р' = p/а. При возникновении лиофильных коллоидных систем с достаточно малой концентрацией частиц дисперсной фазы, когда число частиц oV, много меньше числа молекул дисперсионной среды 2, р « 15-30. Таким образом, величина ос зависит от размера и концентрации образующихся частиц дисперсной фазы и для частиц радиусом ~ КГ8 м составляет при комнатной температуре десятые или сотые доли мДж/м2. Поскольку критериальное значение ос зависит от размера частиц d, образование дисперсной системы с более крупными частицами возможно только при более низких значениях а.
Если дисперсия самопроизвольно возникает из макрофазы при а < ас (и не обнаруживает при этом тенденции к дальнейшему дроблению частиц до отдельных молекул), то она является термодинамически устойчивой. Ребиндер предложил называть подобные дисперсии лиофильными коллоидными системами. Для лиофильных коллоидных систем характерно равновесное распределение частиц по размерам, которое не зависит от пути их возникновения — при диспергировании макроскопической фазы или в результате агрегирования молекул при концентрировании системы.
В противоположность этому лиофобные дисперсные системы, в которых межфазное поверхностное натяжение превышает (обычно на несколько порядков) критическое значение <jc, термодинамически неустойчивы относительно процесса разделения на макроскопические фазы и не могут образовываться самопроизвольным диспергированием макрофазы в отсутствие механических воздействий. Наряду с типичными лиофобными и лиофильными системами могут реализоваться различные промежуточные по природе устойчивости дисперсии, в которых, в зависимости от степени родственности дисперсной фазы и дисперсионной среды, а также концентрации и размера частиц дисперсной фазы, роль теплового движения частиц может быть различной (см. гл. VII).</j
Образование равновесной коллоидно-дисперсной системы возможно при условии, что значение dm лежит в той области дисперсности, где размер частиц заметно превышает молекулярные размеры Ь, т. е. dm » Ь, например, в области dm ? (5 - 10)1». Тогда условие самопроизвольного образования лиофильной коллоидной системы из макрофазы и, соответственно, ее термодинамической равновесности относительно этой макрофазы можно записать в виде критерия, сформулированного Ребиндером и Щукиным:
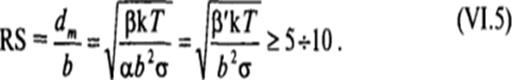
Этот критерий эквивалентен условию самопроизвольного диспергирования макрофазы (VI.4). Таким образом, при достаточно низких, но конечных положительных значениях а, когда а$ае,т. е. при RS » 5 — 10, могут самопроизвольно путем диспергирования макрофазы возникать термодинамически равновесные лиофильные дисперсные системы с заметной концентрацией частиц дисперсной фазы, существенно превосходящих молекулярные размеры.
Условие самопроизвольного образования лиофильной дисперсной системы и ее равновесия относительно макрофазы можно получить также с позиций теории флуктуации. Этот случай удобно рассматривать на примере образования дисперсной системы с легкоподвижной границей раздела фаз (жидкость — жидкость, жидкость — пар). Поверхность жидкости не является абсолютно плоской: в результате термических флуктуаций на ней возникают так называемые капиллярные волны. Как показано Л.И. Мандельштамом (1914), вблизи критической температуры, например абсолютного смешения двух жидкостей, поверхность раздела фаз приобретает четко выраженную шероховатость, что проявляется, в частности, в резком усилении рассеяния света, отраженного от такой поверхности (рис. V1-2). Работу флуктуационного образования на
поверхности единичного «бугорка* (например, в форме полусферы радиусом г, которая затем обособится как отдельная капля) можно выразить как

Рис. VI-2. Отражение света от межфазной поверхности вблизи критической температуры смешения

В соответствии с общей теорией флуктуации (смл;л. V.2) среднее значение квадрата радиуса г2 таких флуктуаций определяется второй производной работы флуктуации по флуктуирующему параметру, т. е. по радиусу г:
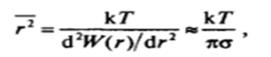
откуда,

Это выражение аналогично условию (VI .4) и отличается от него только численным коэффициентом. Однако оценки о( на основании (VI.6) дают заниженные результаты, поскольку это уравнение не учитывает ряд других факторов, таких, как время ожидания (частоту) флуктуации данного размера и соответственно число (концентрацию) таких частиц.
Поскольку величина р, определяемая выражением (VI. 1), падаете увеличением числа частиц дисперсной фазы в рассматриваемой системе, глубина минимума A#tnjn будет уменьшаться по мере увеличения gVj. Равновесное число частиц радиусом г в единице объема лиофильной коллоидной системы п(г) приближенно определяется условием Afcun = О (ПРИ более точном рассмотрении следует анализировать условие минимума свободной энергии системы при изменении числа частице ней <7Vi). Эта величина, описываемая соотношением
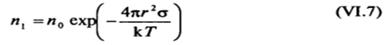
(л0приближно равно числу молекул дисперсионной среды в единице объема системы), может рассматриваться как «коллоидная растворимость» вещества дисперсной фазы в виде частиц радиусом г. Общую коллоидную растворимость можно определить суммированием выражений вида (VI.7) для частиц всех возможных размеров. Чтобы получить коллоидную растворимость, выраженную в количестве вещества, находящегося в дисперсном состоянии в единице объема системы, необходимо учитывать массу частиц разного размера, т. е. при суммировании умножать (VI.7) на число молей вещества в частице радиусом г. Поскольку коллоидная растворимость является экспоненциальной функцией поверхностного натяжения, ее значение может изменяться в широких пределах. Концентрация насыщения частицами коллоидного раствора в случае весьма малых значений о < <тс может быть достаточно большой, тогда как при обычных значениях а > ас оказывается ничтожной (при заданном размере частиц).
Таким образом, рассмотренные представления в наиболее общем виде отражают специфику коллоидно-дисперсного состояния; они включают в термодинамическое описание дисперсной системы два слагаемых, различных по природе и соизмеримых для коллоидных дисперсий по порядку их величины: работу диспергирования и энтропийный выигрыш, связанный с участием частиц в броуновском движении (т. е. теплоту, получаемую в изотермическом процессе из окружающей среды).
Введение концентрации частиц дисперсной фазы как самостоятельной переменной сближает описание термодинамических свойств коллоидных систем и молекулярных (истинных) растворов, т. е. микрогетерогенных и гомогенных систем. Промежуточное положение коллоидно-дисперсных систем между типичными гетерогенными системами, включающими макрофазы, и гомогенными растворами приводит к тому, что по мере роста дисперсности частиц дисперсной фазы становятся все более существенными характерные особенности молекулярно-дисперсного состояния вещества и постепенно ослабевает роль свойств дисперсных систем, роднящих их с макрофазами. Так, грубодисперсным системам свойственно наличие хорошо сформированной поверхности раздела фаз, к которой может быть отнесена поверхностная энергия. Частицы в таких системах содержат достаточно большое число молекул, чтобы можно было говорить об их статистических (усредненных) свойствах. Вместе с тем, уже в этих системах возникают характерные отличия свойств частиц от макроскопических фаз: химический потенциал вещества дисперсной фазы начинает зависеть от размера частиц (см. 1.5).
При увеличении дисперсности системы, когда размер частиц оказывается соизмеримым с толщиной поверхностного слоя, понятие поверхностной энергии, а следовательно и величины о, оказывается более условным. Характерным свойством таких высокодисперсных (коллоидных) систем становится возрастающая роль участия частиц дисперсной фазы в тепловом движении, т. е. статистический характер совокупности большого числа образующих систему частиц. Согласно представлениям Хилла[2], для коллоидного состояния наиболее характерен этот переход от статистических свойств молекул, образующих одну частицу, к статистическим свойствам совокупности коллоидных частиц.
Такое совмещение черт, присущих двухфазным и однофазным системам, позволяет рассматривать дисперсные системы с разных точек зрения. Дисперсии можно считать двухфазными системами с некоторыми особыми свойствами, а именно, химический потенциал вещества дисперсной фазы зависит от дисперсности и в рассмотрение должна вводиться энтропия смешения частиц с молекулами дисперсионной среды. С другой стороны, высокодисперсную (несвязную!) систему можно условно трактовать и как однофазный коллоидный раствор с крупными молекулами-частицами. В этом случае поверхностную энергию одного моля части и (6 ? 10” части ц) можно рассматривать как свободную энергию их «растворения*. Такое смыкание понятий (поверхностная энергия — теплота растворения, дисперсная система — раствор и др.) при переходе от макрофаз к дисперсным и коллоидно-дисперсным системам, а затем к истинным растворам служит яркой иллюстрацией того, как накопление количественных изменений системы диалектически приводит к возникновению качественно новых ее состояний и описывающих эти состояния понятий.
Существенной особенностью дисперсного состояния вещества является и отмеченная Хиллом неоднозначность определения химического потенциала вещества дисперсной фазы. В самом деле, рассмотрим большую по объему дисперсную систему, содержащую «один моль» частиц радиусом г. Избыточный химический потенциал вещества дисперсной фазы, рассматриваемый с соответствующим знаком как работа обратимого изотермического переноса моля вещества из системы в макрофазу, может быть определен в этом случае двумя принципиально различными способами. Можно от каждой частицы отнять по одной молекуле, оставив тем самым неизменным число частиц, но изменив их размер, либо, наоборот, изъять из системы AN = IVJAn/* частиц, в которых содержится моль вещества, не изменив размера остальных частиц. Первый способ определения изменения химического потенциала Др, не включает учета изменения энтропии в образовании дисперсной системы и тождествен с рассмотренным в гл. 1.5. Напротив, второй подход существенным образом учитывает в величине Др роль энтропийных эффектов. Дифференцируя выражение (VI.3) с учетом (VI.2) по числу молей V, при г - const, получаем

Если в системе присутствуют частицы разных размеров, то при малых значениях oVj в этом выражении Щ = i?Vj(г), а произведение 2NA = — общее число кинетически независимых единиц в системе, т. е. молекул растворителя и частиц дисперсной фазы всех размеров, включая отдельные растворенные молекулы вещества дисперсной фазы.
Эти два подхода к определению избыточного химического потенциала вещества дисперсной фазы Др, и Др', используют для анатиза различных аспектов состояния равновесия дисперсной системы. Первый из них был применен в гл. 1.5 к рассмотрению равновесия частицы дисперсной фазы со средой при выводе уравнения Томсона (Кельвина). Второй подход, учитывающий участие частиц в тепловом движении, предусматривает тем самым появление и исчезновение частицы как целого и позволяет описать равновесие частиц различного размера в дисперсной системе. Равновесному распределению частиц по размерам отвечает условие постоянства химического потенциала для частиц различного размера (включая и молекулярные), т. е. Др', - const. Из соотношения (VI.8) получаем выражение для равновесного числа частиц данного радиуса г:
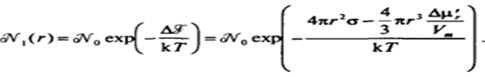
Переходя к единице объема дисперсной системы, это выражение можно записать в виде
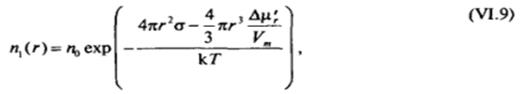
где л, и п0 — число частиц дисперсной фазы и общее число кинетически независимых единиц в единице объема системы соответственно[3].
Таким образом, равновесное содержание частиц и их распределение по размерам описывается больцмановским распределением по значениям свободной энергии. Здесь величина Др'г характеризует степень «насыщенности* системы дисперсной фазой или степень ее метастабильности: при Дц', = 0 система насыщена, при Дц', < 0 — недо- сыщена и при Др', > 0 — пересыщена относительно макрофазы.
В последующих параграфах этой главы будут рассмотрены условия возникновения, строение и характерные свойства лиофильных коллоидных систем, прежде всего систем, содержащих поверхностно-активные вещества, а затем закономерности конденсационного образования лиофобных дисперсных систем, особенности их получения методами механического диспергирования.
Дата добавления: 2019-07-15; просмотров: 217; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!