MUSCLE (Итеративное выравнивание).
Биоинформатика: зачет
Биоинформатика.
Биоинформатика — это междисциплинарная область, которая разрабатывает методы и программные средства для понимания биологических данных. В качестве междисциплинарной области науки биоинформатика сочетает компьютерную науку, статистику, математику и технику для анализа и интерпретации биологических данных.
Составные части биоинформатики:
- 2D и 3D биология
- Разработка биологических баз данных
- Генетические сети и их использование
- Геномика
- Протеомика
- Рентгеноструктурный анализ (РСА) макромолекул
- Индикаторы качества модели макромолекулы, построенной по данным РСА
- Алгоритмы вычисления поверхности макромолекулы
- Алгоритмы нахождения гидрофобного ядра молекулы белка
- Алгоритмы нахождения структурных доменов белков
- Пространственное выравнивание структур белков
- Структурные классификации доменов SCOP и CATH
- Молекулярная динамика
Информация.
Информация - сведения об окружающем мире и протекающих в нем процессах, воспринимаемые человеком или специальным устройством.
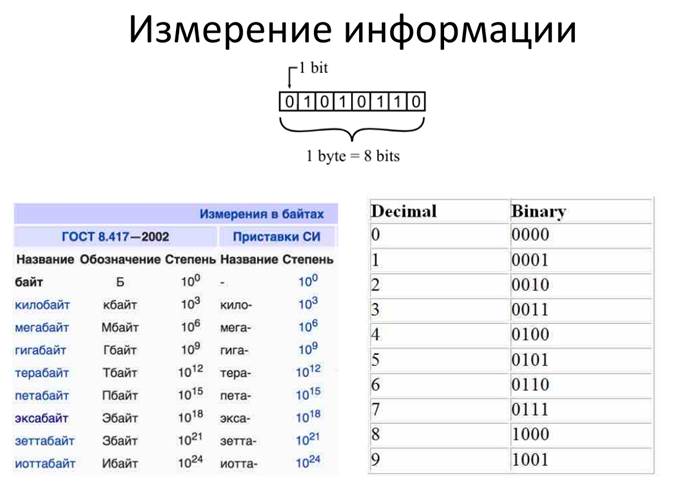
Информационная энтропия
Клод Шеннон (создатель информационной теории) предположил, что прирост информации равен утраченной неопределённости, и задал требования к её измерению:
1.мера должна быть непрерывной; то есть изменение значения величины вероятности на малую величину должно вызывать малое результирующее изменение функции;
|
|
|
2.в случае, когда все варианты (буквы в приведённом примере) равновероятны, увеличение количества вариантов (букв) должно всегда увеличивать значение функции;
3.должна быть возможность сделать выбор (в нашем примере букв) в два шага, в которых значение функции конечного результата должно являться суммой функций промежуточных результатов.
Информацию можно сжимать, хранить, передавать, анализировать, редактировать и т.д. и т.п.
Передача информации
• Комплексная область: теоретические, практические, физические аспекты
• Вопросы сжатия данных
• Вопросы надежности
• Вопросы шифрования и защиты данных (особенно в медицине и биологии).
Передача информации - связь частоты сигнала и пропускной способности.
Теорема Котельникова-(Найквиста Шенона): "любую функцию F(t), состоящую из частот от 0 до f, можно непрерывно передавать с любой точностью при помощи чисел, следующих друг за другом через 1/(2f) секунд".
Теорема Шеннона — Хартли: пропускная способность канала C C {\displaystyle C}, означающая теоретическую верхнюю границу скорости передачи данных, которые можно передать с данной средней мощностью сигнала S S {\displaystyle S} через аналоговый канал связи, подверженный аддитивному белому гауссовскому шуму мощности N N {\displaystyle N} равна:
|
|
|

Базы данных.
База данных — совокупность данных, организованных для удобного поиска и обработки хранимой информации с помощью ЭВМ. Наиболее крупные базы данных, где можно найти информацию о нуклеотидных последовательностях ДНК: GenBank, DDBJ, ENA SRA, Ensembl, dbSNP.
Базы данных для биологии:
• Бесплатные vs Платные (по подписке)
• Свободно доступные vs Ограниченно доступные
• Большие ресурсы (NCBI, EBI/EMBL, etc.) интегрирующие многие базы данных - поддерживаются государстом
• Коллаборации между университетами (напр. PDB)
• Коммерческие компании
• Локальные базы данных, поддерживаемые силами научных групп
• База данных vs Web Server – граница размыта.
• Хорошие БД - информационные ресурсы с возможностями сложного поиска и моделирования.
Что храниться?
• БД статей, абстрактов, патентов: PubMed, Embase, Google Scholar и др.
• Последовательности ДНК (Нуклеотидные БД – это хранилища, принимающие данные от научного сообщества и представляющие их широкой общественности): GenBank, RefSeq.
• Последовательности белков: UniProtKB .
• 3D структуры молекул: PDB (содержит информацию об экспериментально определенных структурах белков, нуклеиновых кислот и различных комплексов), NDB (для сбора и распространения информации о структуре нуклеиновых кислот).
• Геномы: NBCI , e ! Ensembl и др.
• Данные экспрессии: GEO , Exspression Atlas.
• Сырые данные с секвенаторов: SPA (NBCI)
• Информация о химических соединениях и их активности: PubChem, CAS.
• Информация о болезнях, информация о пациентах: ClinicalTrials . gov и др.
• Информация о видах живых организмов: Taxonomy Browser , AlgaeBase , ("Ноев Ковчег")
• Информация о метаболических и сигнальных путях: KEGG PATH WAY, Metacore и др.
• Информация о взаимодействии молекул: STRING, MINT , Kalium и др.
• Много производной информации: базы гомологичных последовательностей, аннотация отдельных классов белков и т.д.
|
|
|
Точечная матрица сходства.
Точечная матрица гомологии (ТМГ, дот-матрица):
Если последовательности близки друг другу и содержат относительно небольшое число делеций то выравнивание может быть получено с помошью карт точечной гомологии или дот-матриц. При этом на графическом устройстве вывода одна последовательность располагается горизонтально, а другая вертикально и на плоскость наносятся точки если в строке и колонке в данной позиции стоит один и тот же символ. Если две последовательности идентичны, то вы увидите сплошную диагональ из точек. В зависимости от сходства последовательностей диагональный элемент будет выражен сильнее или слабее. Делеции видны по сдвигу частей диагонали вправо или влево.
|
|
|
Однако ясно, что при длине последовательности превышающей разрешение графического устройства в несколько раз вся картинка будет полностью зарисована точками. Для того чтобы этого не происходило, а также для большей четкости картинки, применяют разнообразные фильтры. Например, последовательности разбиваются на под-слова длинны n и точка в позиции i,j ставится только тогда, когда в двух под-словах начинающихся в позициях i и j соответвственно совпадает не менее k символов.


Меры сходства последовательностей:
Расстояние по Хэммингу (1950) – количество несовпадающих позиций между последовательностями одинаковой длины;
Расстояние по Левенштейну («редакционное расстояние») (1965) – минимальное число операций редактирования, необходимых для превращения одной строки в другую (длины строк могут не совпадать).
Однако для более аккуратной оценки нужно иметь в виду, что:
- Некоторые замены происходят вероятнее других (особ. аминокислотные);
- Совместная делеция нескольких остатков более вероятна, чем независимая;
- и др....
С этих мер и начинается выравнивание последовательностей. Нам же нужно понять, что такое мы отсеквенировали, какие у него свойства и с чем его едят)
Матрицы замен:
Матрица замен PAM( принцип работы, отличия разных):
Аминокислоты с близкими биохимическими свойствами, такими как заряд, полярность и т.д. характеризуются большей вероятностью парных замен. Некоторые аминокислоты, например цистеин, глицин, триптофан очень редко заменяются в процессе эволюции. Для того чтобы учесть неравную вероятность замен были разработаны специальные матрицы, которые получили название матрицы замен. Эти матрицы содержат оценки частных весов для любой пары замены аминокислоты (или нуклеотида) i на аминокислоту (или нуклеотид) j.
Accepted Point Mutation (APM->PAM) – замена одной аминокислоты на другую, закрепившаяся в процессе эволюции.
PAM (Percent Accepted Mutation) – единица измерения расхождения последовательностей в выравнивании.
РАМ - самая первая матрица замен, созданная для аминокислот. В ней действует следующее правило:
- Если вес S(i, j) в ячейке i, j таблицы 1 больше нуля означает, что аминокислота i заменяется на j чаще, чем в среднем по всем заменам. То есть эти аминокислоты сравнительно легко заменяют друг друга, т.к. они функционально эквивалентны или по другим причинам.
- Если вес меньше нуля указывает на пары аминокислот, которые сравнительно редко заменяют друг друга.
Матрицы PAM различаются по числовым индексам. PAMN – набор матриц вероятностей замен, аппроксимированных для выравниваний, содержащих N замен на 100 остатков. Исходно матрица PAM1 нормирована из расчета не более 1 мутации на 100 остатков; остальные получаются матричным умножением. Например, матрице PAM250, соответствует примерно 20 % идентичности последовательностей, что считается минимальным уровнем сходства, для которого можно надеяться получить правильное выравнивание, основываясь на анализе самих последовательностей без привлечения дополнительной информации, например, пространственной организации белковой глобулы. Расстояние 250 PAM означает, что при эволюции последовательности длиной 100 аминокислотных остатков произошло 250 мутаций в случайных позициях. Поэтому в некоторых позициях мутаций вообще не было, а в некоторых позициях произошло 3 и более мутационных изменений.
Недостатком матриц РАМ является то, что они не очень надежно работают на больших эволюционных расстояниях (слишком много мутаций).
Другие матрицы:
Другим широко используемым семейством матриц весов являются матрицы BLOSUM, предложенные в 1992 г. Они построены на основе выравниваний последовательностей с определенной степенью сходства.
Принцип такой: в матрицах BLOSUMзначение веса S (i, j) для каждой ячейки i, j получено из наблюдений частот замен в частичных выравниваниях близких белков. Каждая матрица соответствует специфическому порогу сходства. Например, при построении матрицы BLOSUM62 были использованы последовательности, имеющие более чем 62% сходства.
Матрицы с меньшими пороговыми значениями соответствуют большим временам раздельной эволюции. Поэтому их используют для выравнивания более удаленных друг от друга последовательностей.
Основными отличиями матриц РАМ и Blosum являются:
1) использование матрицами РАМ простой эволюционной модели (подсчет замен на ветвях филогенетического древа);
2) матрицы РАМ основаны на учете мутаций по принципу глобального выравнивания (в высококонсервативных и высокомутабельных участках), а матрицы Blosum – локального (только высококонсервативных участков).
Так же используются матрицы Gonnet, представляющие собой усовершенствованный вариант матриц Дэйхофф, основанный на большей базе данных.
Маргарет Дэйхоф (1925 – 1983):
- Инициировала создание Atlas of Protein Sequence and Structure (1965)
- Первой начала использование компьютеров для сравнения последовательностей
- Предложила однобуквенный код для аминокислот
- Впервые провела реконструкцию эволюционного дерева исходя из выравнивания последовательностей (1966)


Метод динамического программирования:
Динамическое программирование в теории управления и теории вычислительных систем — способ решения сложных задач путём разбиения их на более простые подзадачи. Он применим к задачам с оптимальной подструктурой, выглядящим как набор перекрывающихся подзадач, сложность которых чуть меньше исходной. В этом случае время вычислений, по сравнению с «наивными» методами, можно значительно сократить.
Ключевая идея в динамическом программировании достаточно проста. Как правило, чтобы решить поставленную задачу, требуется решить отдельные части задачи (подзадачи), после чего объединить решения подзадач в одно общее решение. Часто многие из этих подзадач одинаковы. Подход динамического программирования состоит в том, чтобы решить каждую подзадачу только один раз, сократив тем самым количество вычислений. Это особенно полезно в случаях, когда число повторяющихся подзадач экспоненциально велико.
Динамическое программирование сверху — это простое запоминание результатов решения тех подзадач, которые могут повторно встретиться в дальнейшем.
Динамическое программирование снизу включает в себя переформулирование сложной задачи в виде рекурсивной последовательности более простых подзадач.
Алгоритм Нидлмана-Вунша:
Алгоритм Нидлмана - Вунша — это алгоритм для выполнения выравнивания двух последовательностей, который используется в биоинформатике при построении выравниваний аминокислотных или нуклеотидных последовательностей. Алгоритм был предложен в 1970 году Солом Нидлманом и Кристианом Вуншем
Этот алгоритм является примером динамического программирования (способом решения сложных задач путём разбиения их на более простые подзадачи), и он оказался первым примером приложения динамического программирования к сравнению биологических последовательностей. Используется для глобального выравнивания 2 последовательностей.
Принцип алгоритма:
Суть алгоритма в следующем. По матрице расстояний между аминокислотами (или, соответственно, между нуклеотидами) итеративным образом рассчитывается матрица всех возможных маршрутов *Sij*: Sij=Dij+max(Si-1,j-1,max k<j-1 (Si-1,k - G),max k<i-1 (Sk,j-1-S)) где Sij - элемент i-й строки j-го столбца, Dij - расстояние между i-й и j-й аминокислотами (или нуклеотидами), а G - штраф на делецию. Затем осуществляется проход по матрице в обратном направлении, по максимальным элементам. Полученный маршрут соответствует оптимальному выравниванию. В качестве матрицы минимальных расстояний между аминокислотами обычно используется матрица минимальных мутационных расстояний по генетическому коду между аминокислотами, но могут использоваться и другие меры.
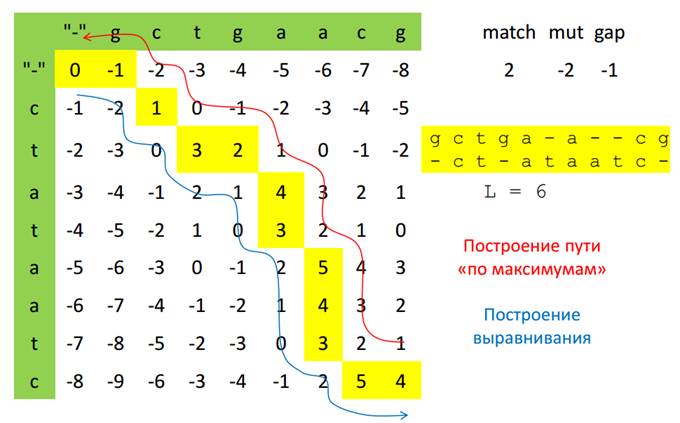
Экспресс-метод сравнения последовательностей - BLAST:
BLAST - (Basic Local Alignment Search Tool (Altschul et al.,1990)) предназначен для сравнения новых последовательностей с уже содержащимися в базах данных.
Алгоритм:
- Удаление малоинформативных участков последовательности (повторы и т.п.);
- Составление списка k-буквенных слов (K-tuple), присутствующих в последовательности запроса;
- Сопоставление этих слов со всеми возможными словами длины к и оценка сходства; отбор слов с оценкой, превышающей пороговую (например, для слова PQG сходными будут PNG, PEG и PDG, но не PQW).
- Сканирование последовательности из БД и поиск в ней слов c высокой оценкой, полученных на предыдущем шаге;
- Расширение локальных выравниваний в обе стороны до тех пор, пока суммарная оценка выравнивания не начинает уменьшаться (построение сегментных пар (high-scoring segment pair, HSP));
- Объединение сегментных пар, лежащих на удалении меньше А;
- Составление списка сегментных областей с высокой оценкой;
- Расчет статистической значимости этих оценок.
Значимость выравнивания:
Определяется по E-value: E < 0,02 высокая вероятность гомологии
0,02 < E < 1 гомология не очевидна
E > 1 сходство случайно
Хотя для коротких последовательностей сходство может быть НЕ случайным даже при E > 1 !! (примерно как с p-value и маленькими выборками).
Распределение нормализованных максимальных оценок HSP подчиняется распределению Гумбеля (распределению экстремальных значений), для которого
P(S n,m > S) ×m º E - value (хз зачем, но пусть будет).
Значимость выравнивания.
Значимость выравнивания:
1. Для подтверждения гомологичости
последовательностей.
2. Если открыта новая последовательность с
неизвестной функцией, но при этом в базах данных могут быть найдены
подобные ей последовательности с ранее установленными структурами и
функциями, то результаты выравнивания (сравнения) этой новой
последовательности с уже исследованными последовательностями могут стать
основанием для предсказания функции или структуры этой новой
последовательности.
3. Для подтверждения новизны отсеквенированного фрагмента (может он уже давно в какой-нибудь БД валяется)
Множественное выравнивание и алгоритмы (Clustal, Muscle).
CLUSTAL (Прогрессивное вы равнивание):
Clustal (1988) выполняет постепенное выравнивание все новых последовательностей, начиная с наиболее <эволюционно> близких, ориентируясь на предварительно построенное на основании парных выравниваний филогенетическое дерево.
Алгоритм:
1) Экспресс-оценка сходства двух последовательностей вычисляется как число совпадающих остатков в словах длины K (K = 1-2 для белковых последовательностей и 2-4 для нуклеотидных) за вычетом штрафа за сделанные вставки.
2) Методом UPGMA (позже NJ) рассчитывается направляющее дерево, по которому затем рассчитываются веса последовательностей, причем более близкие последовательности получают меньшие веса.
3) Согласно направляющего дерева выбираются наиболее близкие последовательности и выполняется их выравнивание методом динамического программирования с использованием матрицы замен и штрафов за открытие/расширение вставок, с полученным выравниванием сопоставляются всё новые последовательности.
Особое внимание уделено значениям штрафов за вставки. Введена их зависимость от:
А) типа сопоставляемого и предшествующего остатков;
Б) степени близости последовательностей;
В) длин рассматриваемых последовательностей;
Г) наличия вставок в уже имеющемся выравнивании;
Д) характера аминокислотной последовательности.
MUSCLE (Итеративное выравнивание).
Итеративное выравнивание - набор методов для построения множественных выравниваний, в которых происходит снижение ошибок, наследуемых в прогрессивных методах. Они работают аналогично прогрессивным методам, но при этом неоднократно перестраивают исходные выравнивания при добавлении новых последовательностей. В отличие от прогрессивных методов, итеративные методы могут возвращаться к первоначально посчитанным парным выравниваниям и подвыравниваниям, содержащим подмножества последовательностей из запроса, и таким образом оптимизировать общую целевую функцию и повышать качество.
Алгоритм включает в себя три стадии:
1) быстрое «черновое» множественное выравнивание (попарные глобальные выравнивания; оценка сходства как доля совпадающих позиций; построение направляющего дерева методом UPGMA или NJ; прогрессивное выравнивание)
2) улучшенное множественное выравнивание (оценка сходства как доля совпадающих позиций в текущем множественном выравнивании; построение направляющего дерева через построение матрицы расстояний по Кимуре и ее кластеризацию; сравнение текущего дерева с построенным ранее; пересчет выравнивания для отличающихся узлов; повторение до сходимости)
3) уточнение выравнивания (удаление произвольного узла для разбиения дерева на два; построение профилей для каждого поддерева и их выравнивание; расчет суммы парных оценок в получающемся множественном выравнивании; перебор всех узлов от листьев к корню ивыбор выравнивания с максимальной суммой).
Методы кластеризации: UPGMA и NJ.
UPGMA (Unweighted Pair Group Method with Arithmetic mean) (1958) – метод невзвешенной группировки с арифметическим средним – пример алгоритма иерархической кластеризации.
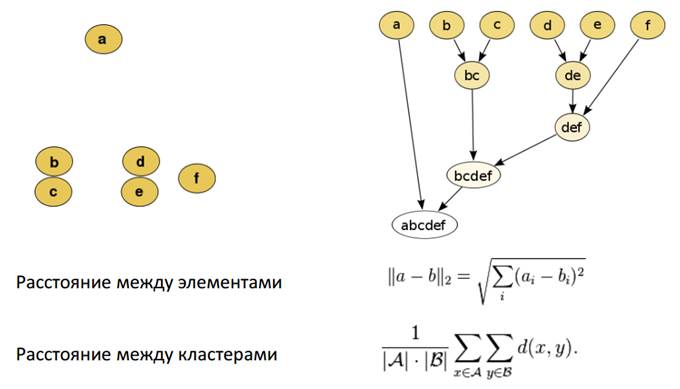
Примерный алгоритм:
Дан набор объектов Sk, где для каждой пары (Si, Sj) установлена мера сходства L(Si, Sj). Для построения дерева выбирают два наиболее близких объекта (Sm, Sn) и добавляют вершину, изображающую их общего «предка» (Smn). Затем замещают эти два объекта группой, содержащий обоих, и присваивают расстояниям от этой пары до остальных объектов Sk средние значения от каждого из элементов этой группы до Sk:
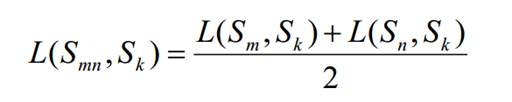
В случае объединения кластеров C_i и C_j с образованием кластера C_k, содержащего n_i + n_j = n_k элементов, расстояние от кластера C_k достальных кластеров C_m вычисляется как:
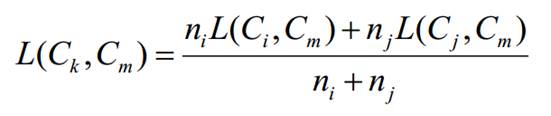
Насколько я поняла - находим самое маленькое значение, а потом ищем средние арифметические между группами значений. Среднее арифметическое будет длиной ветки дерева.
Пример из презы:
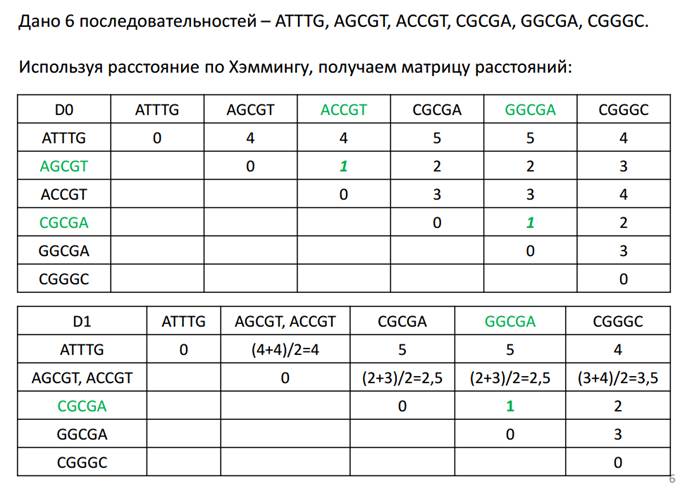
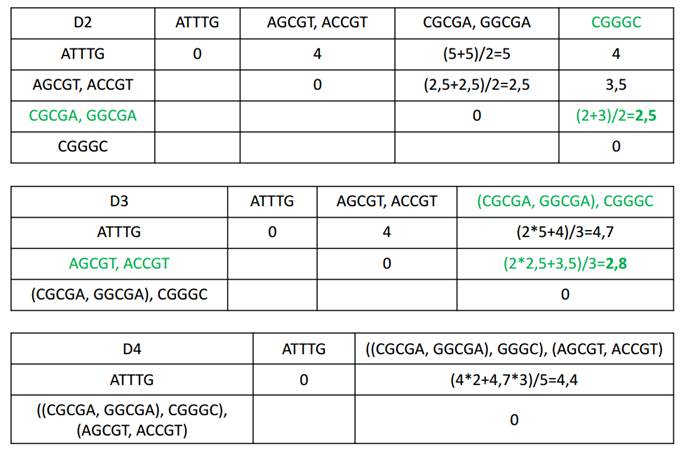
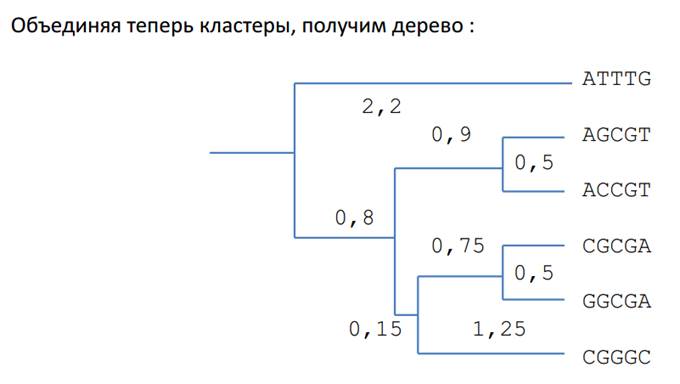
Длины ветвей установлены так, что расстояние от корня одинаково для всех листьев - ультраметричность.
Метод UPGMA подразумевает справедливость гипотезы молекулярных часов (постоянной скорости эволюции).
(Хрень какая-то, но ладно..).
Mетод присоединения соседей, N J (Neighbor joining) —это восходящий кластерный метод для создания филогенетических деревьев. Обычно используется для деревьев, основанных на ДНК или белковых последовательностях. Для его реализации необходимо вычислить расстояния между каждой парой таксонов (например, видов или последовательностей).
Алгоритм :
- Вычисляется матрица попарных расстояний между таксонами.
- По текущей матрице расстояний считается Q {\displaystyle Q} Q-матрица.
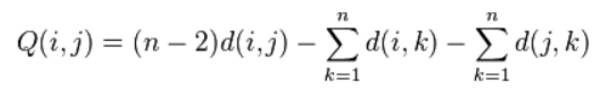
- Ищется пара различных таксонов i {\displaystyle i} i и j {\displaystyle j} j (то есть i ≠ j {\displaystyle i\neq j} i не равно j), для которых значение Q ( i , j ) {\displaystyle Q(i,j)}Q(i,j) — наименьшее. Эти таксоны присоединяются к новому узлу, который, в свою очередь, соединяется с центральным узлом.
- Рассчитывается расстояние от каждого из присоединенных таксонов до нового узла.
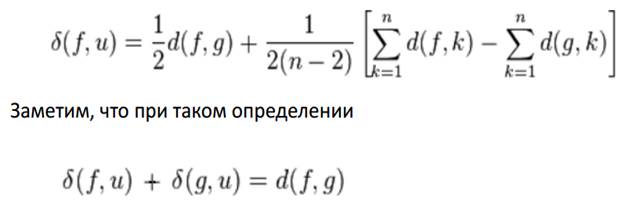
- Рассчитывается расстояние от каждого из оставшихся таксонов до нового узла.
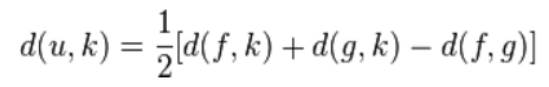
- Формируем новую матрицу попарных расстояний: из текущей матрицы удаляем строки и столбцы, соответствующие только что присоединенным таксонам и добавляем новую вершину и, вычисленные в 5 пункте, расстояния.
- Повторяем шаги 2 — 5, пока дерево не станет полностью разрешённым и не станут известны длины всех ветвей.
Цель: найти такие таксоны, чтобы сумма всех ветвей дерева была минимальна.
Пример смотрите в 7 лекции (слайды 20-29). Но получается вот примерно такая хрень:
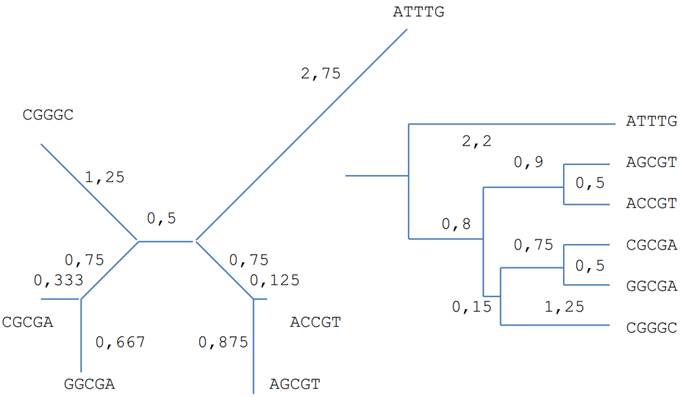
Профиль множественного выравнивания:
Множественное выравнивание последовательностей — выравнивание трёх и более биологических последовательностей, обычно белков, ДНК или РНК. В большинстве случаев предполагается, что входной набор последовательностей имеет эволюционную связь. Используя множественное выравнивание, можно оценить эволюционное происхождение последовательностей, проведя филогенетический анализ.
Визуальное представление выравнивания иллюстрирует мутационные события как точечные мутации в виде различающихся символов в одной колонке выравнивания, а также их вставки и делеции (хромосомные перестройки, при которых происходит потеря участка хромосомы)
Множественное выравнивание последовательностей часто используется для оценки консервативности доменов белков, третичных и вторичных структур и даже отдельных аминокислотных остатков или нуклеотидов.
Ввиду большей вычислительной сложности по сравнению с парным выравниванием множественное выравнивание требует более сложные алгоритмы. Многие соответствующие программы используют эвристические алгоритмы, поскольку поиск глобального оптимального выравнивания для многих последовательностей может занимать очень большое время.
Что полезного?
- Выявление удаленной гомологии
- Выявление консервативных остатков и мотивов
- Построение филогенетических деревьев и т.д.
Алгоритмы:
- Динамическое программирование – не годится!
- Прогрессивное выравнивание
- Итеративное выравнивание
- Скрытые марковские модели
- Квантовые компьютеры (?! (2017))
Визуализация: Построение профилей
Филогенетические деревья, бутстреп:
Филогенетическое дерево (эволюционное дерево, дерево жизни) — дерево, отражающее эволюционные взаимосвязи между различными видами или другими сущностями, имеющими общего предка.
Дерево – связный граф, содержащий только один путь между двумя вершинами.
Вершины филогенетического дерева делятся на три класса: листья, узлы и (максимум один) корень. Листья — это конечные вершины, то есть те, в которые входят ровно по одному ребру; каждый лист отображает некоторый вид живых организмов (или иной объект, подверженный эволюции, например, домен белка). Каждый узел представляет эволюционное событие: разделение предкового вида на два или более, которые в дальнейшем эволюционировали независимо. Корень представляет общего предка всех рассматриваемых объектов. Ребра филогенетического дерева принято называть «ветвями».
Типы деревьев:
· Укоренённое дерево — дерево, содержащее выделенную вершину — корень. Укоренённое дерево можно считать ориентированным графом, поскольку на нём имеется естественная ориентация — от корня к листьям.
· Неукоренённое дерево не содержит корня и отражает связь листьев без предполагаемого положения общего предка.
· Кладограмма — филогенетическое дерево, не содержащее информации о длинах ветвей.
· Филограмма — филогенетическое дерево, содержащее информацию о длинах ветвей; эти длины представляют изменение некой характеристики.
o Хронограмма — филограмма, длины ветвей в которой представляют эволюционное время.
Дендрограмма — общий термин, обозначающий схематическое представление филогенетического дерева.
Филогенетические деревья – методы построения:
- Максимальная экономия (Fitch, 1971) (метод оценки!) – критерий оптимальности, согласно которому предпочтительнее деревья с меньшим суммарным числом мутаций. Однако алгоритма быстрого построения такого дерева не существует.
- Метод максимального правдоподобия учитывает не просто число мутаций, но и их вероятность.
Методы построения деревьев могут быть оценены по нескольким основным показателям:
- эффективность (сколько времени и памяти требуются для вычислений)
- полезность (есть ли польза от полученных данных или информация бесполезна)
- воспроизводимость (будут ли повторные ответы такими же, если каждый раз даются разные данные для той же проблемной модели)
- устойчивость к ошибкам (справляется ли с неточностями в предпосылках рассматриваемой модели)
- выдача предупреждений (будет ли предупреждать нас, когда неправильно используется, то есть предпосылки неверные).
Методы проверки:
1. Использование внешней группы, т.е. видов, которые заведомо более удалены ото всех видов, для которых строится дерево (приматы и корова);
2. Сравнение деревьев, полученных на основе разных характеристик. Очевидно, они должны быть согласованными;
3. Оценка результата с помощью формальных статистических тестов. Например, построение дерева для подмножества последовательностей из исходного множественного выравнивания должно дать поддерево дерева, полученного для этого выравнивания;
4. Бутстреп (boot strap)
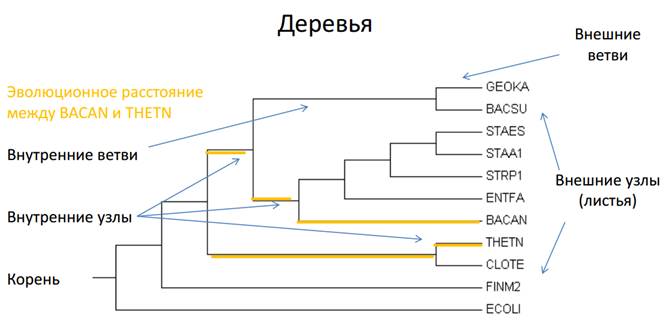
Пример строения дерева:
Бутстрэп в статистике — практический компьютерный метод исследования распределения статистик вероятностных распределений, основанный на многократной генерации выборок методом Монте-Карло на базе имеющейся выборки. Позволяет просто и быстро оценивать самые разные статистики (доверительные интервалы, дисперсию, корреляцию и так далее) для сложных моделей.
Суть метода состоит в том, чтобы по имеющейся выборке построить эмпирическое распределение. Используя это распределение как теоретическое распределение вероятностей, можно с помощью датчика псевдослучайных чисел сгенерировать практически неограниченное количество псевдовыборок произвольного размера, например, того же, как у исходной. На множестве псевдовыборок можно оценить не только анализируемые статистические характеристики, но и изучить их вероятностные распределения. Таким образом, например, оказывается возможным оценить дисперсию или квантили любой статистики независимо от её сложности. Данный метод является методом непараметрической статистики.
Бутстрэп используется для корректировки смещения, тестирования гипотез, построения доверительных интервалов.
Дата добавления: 2019-02-22; просмотров: 1335; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!