Электронный дополнительный материал
Перитонеальный диализ у младенцев: опыт итальянского реестра педиатрического хронического диализа.
Видал Е 1 , Edefonti , Murer л , Gianoglio Б , Maringhini S , Пекораро С , Sorino Р , Leozappa G , Lavoratti G , Ratsch И.М. , Chimenz R , Verrina Е ; Итальянский реестр детского хронического диализа .
Абстрактно
ЗАДНИЙ ПЛАН:
Хотя хронический перитонеальный диализ (CPD) считается заместительной терапией для детей с конечной стадией почечной недостаточности, многие вопросы сохраняются в отношении рисков и результатов лечения.
МЕТОДЫ:
Мы представляем данные о 84 младенцах, которые начали ДСП в возрасте до 1 года; эти пациенты составляют 12% от общей численности итальянского реестра педиатрического хронического диализа. Мы проанализировали записи пациентов от всех детей, которые были последовательно обработаны CPD в период с 1995 по 2007 год в Италии. Анализ данных о росте проводился только у младенцев с полными ауксиологическими параметрами через 0, 6 и 12 месяцев наблюдения.
РЕЗУЛЬТАТЫ:
Средний возраст в начале ДСП составил 6,9 месяца, вес 6,1 кг и длина 63,6 см. В половине исследования диагноз, приводящий к почечной недостаточности, был врожденной нефроуропатией. У 28% детей была по крайней мере одна ранее существовавшая сопутствующая патология. Средняя оценка стандартного отклонения была -1,65 в начале CPD, -1,82 через 12 месяцев и -1,53 через 24 месяца. Захват роста был зарегистрирован у 50% пациентов во время диализа. Положительная корреляция наблюдалась между продольным ростом и объемом обмена (R (2) = 0,36), так и длиной сеанса диализа (R (2) = 0,35), а отрицательная связь была обнаружена с числом случаев перитонита (P = 0,003) , Частота перитонита была 1: 20,7 эпизода: CPD-месяцы (1: 28,3 у детей старшего возраста из того же реестра) и была значительно выше у детей с олигоанурией (1: 15,5 эпизод: CPD-месяцы) по сравнению с младенцами с остаточной функцией почек (эпизод 1: 37,4: CPD-месяцы). Выживаемость катетера составляла 70% через 12 месяцев и 51% через 24 месяца. Связанные с катетером осложнения были сходными у детей грудного и раннего возраста (1: 20,5 против 1: 19,8 эпизода: CPD-месяцы), тогда как клинические осложнения были более частыми у детей в возрасте до 1 года (1: 18,3 против 1: 25,2 эпизода: CPD -месяцы, P <0,05). В течение периода наблюдения 33 пациента были пересажены (39,3%), 18 перенесены на гемодиализ (21,4%) и 8 умерли (9,5%). Коэффициент смертности был в 4 раза выше, чем у детей старшего возраста (2,3%). Связанные с катетером осложнения были сходными у детей грудного и раннего возраста (1: 20,5 против 1: 19,8 эпизода: CPD-месяцы), тогда как клинические осложнения были более частыми у детей в возрасте до 1 года (1: 18,3 против 1: 25,2 эпизода: CPD -месяцы, P <0,05). В течение периода наблюдения 33 пациента были пересажены (39,3%), 18 перенесены на гемодиализ (21,4%) и 8 умерли (9,5%). Коэффициент смертности был в 4 раза выше, чем у детей старшего возраста (2,3%). Связанные с катетером осложнения были сходными у детей грудного и раннего возраста (1: 20,5 против 1: 19,8 эпизода: CPD-месяцы), тогда как клинические осложнения были более частыми у детей в возрасте до 1 года (1: 18,3 против 1: 25,2 эпизода: CPD -месяцы, P <0,05). В течение периода наблюдения 33 пациента были пересажены (39,3%), 18 перенесены на гемодиализ (21,4%) и 8 умерли (9,5%). Коэффициент смертности был в 4 раза выше, чем у детей старшего возраста (2,3%).
|
|
|
|
|
|
ВЫВОДЫ:
Наши данные подтверждают, что младенцы на CPD представляют группу высокого риска; однако наш опыт показал, что рост был приемлемым, и большая часть была успешно пересажена. Усиленные усилия должны быть направлены на оптимизацию эффективности диализа и предупреждение перитонита. Более высокий уровень смертности у младенцев в значительной степени был вызван сопутствующими заболеваниями.
Источник: https://www.ncbi.nlm.nih.gov/pubmed/21669887
Трансплантация гематопоэтической стволовых клеток у младенца с иммунодефицитом, центромерная нестабильность и синдром лицевой аномалии
|
|
|
Абстрактно
Иммунодефицит, центромерная нестабильность и синдром лицевой аномалии (ICF) представляют собой редкое аутосомно-рецессивное генетическое состояние с тяжелым иммунодефицитом, которое приводит к летальным инфекциям, если их не распознать и лечить в раннем детстве. Современные схемы лечения состоят из профилактического и поддерживающего лечения рецидивирующих инфекций. Здесь мы сообщим о случае 1-летнего мальчика из марокканских родственников, которому был поставлен диагноз 4 месяца с синдромом ICF с гомозиготной мутантной мутацией в гене DNMT3B . Сначала он был госпитализирован с рецидивирующими легочными инфекциями из оппортунистического патогена Pneumocystis jirovecii (PJ), Дальнейшая иммунологическая обработка выявила агаммаглобулинемию в присутствии В-клеток. После успешного выздоровления от пневмонии PJ он перенес трансплантацию гемопоэтических стволовых клеток (HSCT) у здоровой сестры, совместимой с HLA, с использованием химиотерапевтического режима кондиционирования, состоящего из треосульфана, флударабина и тиотепа. Помимо острых побочных эффектов, связанных с химиотерапией, серьезных побочных эффектов не произошло. Через шесть месяцев после восстановления иммунитета HSCT у него был стабильный химеризм с 2,9% аутологичной порцией периферической крови и нормальным дифференциальным числом клеток крови, включая все подтипы иммуноглобулина. Это один из первых случаев успешного HSCT при синдроме ICF. Ранняя диагностика и последующий HSCT могут предотвратить серьезные оппортунистические инфекции и вылечить иммунодефицит. Центровая нестабильность и лицевая аномалия остаются неизменными. Хотя долгосрочный результат пациента и неврологическое развитие еще предстоит увидеть, эта лечебная терапия для иммунодефицита улучшает ожидаемую продолжительность жизни и качество жизни. Этот случай предназначен для повышения информированности врачей о синдроме ICF и на раннем этапе подчеркнуть важность HSCT в синдроме ICF.
|
|
|
Ключевые слова: иммунодефицит, центромерная нестабильность, лицевая аномалия, синдром ICF, агаммаглобулинемия, трансплантация гемопоэтических стволовых клеток, пневмония пневмоцистной пневмонии
Введение
Иммунодефицит, центромерная нестабильность и синдром лицевой аномалии (ICF) являются редким аутосомно-рецессивно унаследованным генетическим состоянием. Большинство затронутых лиц имеют мутации в гене метилтрансферазы 3B ( DNMT3B , OMIM 602900) на хромосоме 20, что приводит к уменьшению метилирования ДНК перицентромерных областей хромосом 1, 9 и 16 ( 1 , 2). Эпигенетическая дисрегуляция, а не один дефект гена, определяет клинический фенотип. Хотя все клетки организма несут ту же самую мутацию, различные ткани по-разному влияют из-за различной степени метилирования ДНК. Это особенно заметно в митоген-стимулированных лимфоцитах, где целые плечевые делеции, транслокации и многоразветвленные хромосомы вызывают аномальную регуляцию генов изотипа изотипа В-клеточного иммуноглобулина, активацию лимфоцитов и миграцию ( 3 ). Пациенты МКФ страдают от рецидивирующих желудочно-кишечных и легочных инфекций в раннем детстве из-за агаммаблобулинемии, что приводит к неспособности развиваться ( 4 ). Внутренний дефект Т-клеток также связан с высокой частотой оппортунистических инфекций от патогенов, таких какPneumocystis jirovecii (PJ) , но точный механизм не был выяснен ( 5 ). Типичные клинические характеристики включают одноименную лицевую аномалию эпикантичных складок, гипертелоризм и плоский носовой мост, а также задержку в психологическом и когнитивном развитии.
Варианты лечения ограничены и состоят в основном из поддерживающей терапии , таких как замещение иммуноглобулинов, профилактической сульфаметоксазол-триметоприм терапии или терапии антибиотиками ( 6 - 8 ). Ожидаемая продолжительность жизни пациентов МКФ плохая, а прогноз зависит от частоты и тяжести инфекций. Высокая доля зарегистрированных пациентов с МКФ умирает в раннем возрасте ( 9 ). Ранняя замена IgG и антибиотикопрофилактика могут значительно улучшить результаты лечения пациентов ( 10 ). Ранняя устойчивая терапия иммунодефицита может значительно улучшить курс болезни. Единственным лечебным лечением иммунной дисфункции является трансплантация гемопоэтических стволовых клеток (HSCT), как описано в отдельных примерах ( 3, 7 ). Однако на сегодняшний день не существует долгосрочных данных о последующих действиях.
Задний план
Здесь мы сообщим о случае 1-летнего мальчика марокканского кровеносного происхождения с диагнозом синдрома ICF, несущего гомозиготную миссенсусную мутацию в гене DNMT3B(Ala603Thr) с гипогаммагобулинемией, нормальным количеством В-клеток, лицевой аномалией и неспособностью процветать.
Этот вариант был ранее описан в синдроме ICF Hansen et al. Он состоит из точечной мутации (rs121908943) и обмена аминокислот от аланина до треонина. Вариант имеет незначительную частоту аллеля A = 0,000008 ( 11 ) и показатель распространенности менее 1/1000000 . Hansen et al. описал этот вариант среди других мутаций в гене DNMT3B, в то время как 105 контрольных ДНК от неродственных здоровых кавказцев в Северной Америке и Нидерландах не несли эту мутацию ( 1).
Мальчик родился с массой тела 3200 г (38-й процентиль) и длиной 51 см (72-й процентиль) (диаграммы роста CDC / WHO 2000). В возрасте 2,5 месяцев его вес тела составлял 5,300 г (12-й процентиль), а длина составляла 59 см (24-й процентиль) (диаграммы роста CDC / ВОЗ, 2000 год). В возрасте 4 месяцев пациент впервые был госпитализирован с повторными и продолжительными респираторными инфекциями нижних и верхних дыхательных путей с гипоксемией. Лабораторная работа выявила двустороннюю бронхопневмонию, положительную для вируса парагриппа 1 и 3 и PJ , а также гнойный конъюнктивит и гнойный средний отит, перфоранс, положительный для Haemophilus influenzae, В последующей иммунологической диагностике, значительно снижаются уровень всех иммуноглобулинов подклассов (таблица (Таблица 1)1 ) и нормальные В - клетки рассчитывать с было обнаружено отсутствие В - клеток памяти. Количество Т-клеток и скорость пролиферации CD4 + и CD8 + клеток после митогенной стимуляции фитогемагглютинином были нормальными. Бронхоскопия выявила нормальные анатомические пропорции дыхательных путей. PJпневмонию лечили внутривенным котримоксазолом, кортикостероидами и кислородными добавками. Последующее регулярное внутривенное введение иммуноглобулинов и профилактический котримоксазол предотвращали дальнейшие тяжелые легочные инфекции. Кроме того, пациент имеет типичную dysmorphisms лица , состоящую из гипертелоризма, плоский носовой мостика, epicanthic складки, и низкий набор уши (рис (Figure1A-изображения1 A-фотографий на лице). Синдром МКФ была клинический подозреваемыми и подтверждена с цитогенетическим анализ показал всю руку делеции, транслокации и multibranched хромосомы из - за нестабильности в центромерной хромосом 1, 9 и 16 (рис (Figure2-кариограмму).2-karyogram). Пациент является пятым ребенком близкородственных марокканских родителей, которые являются двоюродными братьями и сестрами (рис (Figure1B Беспородного).1 B-породным). Три здоровых старших брата (5-, 4- и 2-летние) развивались нормально. ICF у братьев и сестер исключался с нормальным уровнем иммуноглобулинов в крови и нормальной кариограммой. Самая старшая дочь семьи умерла от респираторной недостаточности в возрасте 5 месяцев в Марокко, дальнейшая генетическая или патологическая диагностика не была выполнена после смерти .
Таблица 1
Иммунологические параметры до и через 6 месяцев после гематопоэтической трансплантации стволовых клеток (HSCT).
| До HSCT | 6 месяцев после HSCT | |
| IgG (мг / дл) | 52 | 571 |
| IgA (мг / дл) | <5 | 57 |
| IgM (мг / дл) | <5 | 51 |
| CD4 + (c / мкл) | +1840 | +818 |
| CD8 + (c / мкл) | 450 | 613 |
| CD4 + / CD8 + | 4,1 | 1,3 |
| CD20 + (c / мкл) | 1329 | 588 |
| CD20 + IgD + CD27- (с / мкл) | 1329 | 588 |
| CD20 + IgD-CD27 + (c / мкл) | 0 | 7 |
| CD56 + (c / мкл) | 208 | 148 |
| CD14 + (c / мкл) | +913 | 292 |
| CD66b + CD49d- (c / мкл) | 2040 | 3723 |
| Химеризм (BM) (% аутологичной части) | - | 1-5 |
| Химеризм (кровь) (% аутологичной части) | - | 2,9 |
Иммуноглобулины (Ig) G, A, A измерялись в сыворотке с помощью ELISA. Т-хелперные клетки (CD4 +), цитотоксические Т-клетки (CD8 +), В-клетки (CD20 +), наивные В-клетки (CD20 + IgD + CD27-), В-клетки памяти (CD20 + IgD- CD27 +), NK-клетки (CD56 +), моноциты (CD14 +), нейтрофильные гранулоциты (CD66b + CD49d-) измеряли проточной цитометрией. Анализ химеризма в костном мозге (БМ) и крови проводили с использованием молекулярно-генетических методов или XY-Fish соответственно .
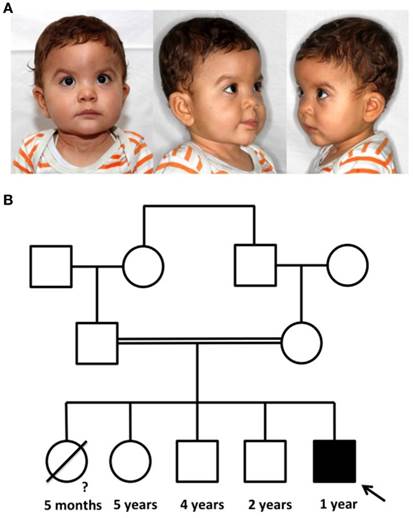
Рисунок 1
Рисунок пациента с иммунодефицитом, центромерной нестабильностью и синдромом лицевой аномалии (ICF) и родословной семьи. (A) Типичными фенотипическими характеристиками синдрома ICF являются эпикантичные складки, телекантус, гипертелоризм, плоский носовой мост и низкорослые и задне вращающиеся уши. (B) Пациент пятый ребенок родственников. Первая дочь умерла в возрасте 5 месяцев от неизвестных причин. Три старших брата и сестры здоровы.
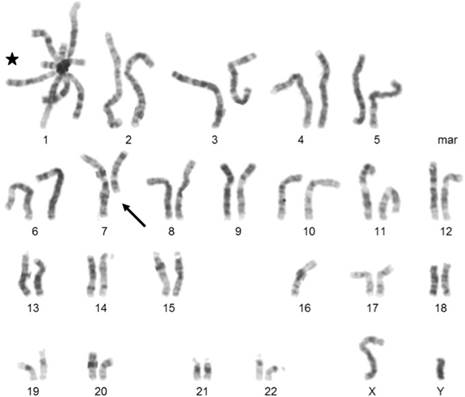
Кариограмма цитогенетического анализа. Эта метафаза показывает образование многорадиальной фигуры (обозначенной звездой), терминальное удаление части длинного плеча хромосомы 7 (обозначено стрелкой) и потерю одной хромосомы 16.
В возрасте 5 месяцев он представил массу тела 6 600 г (8-й процентиль) и длину 66 см (37-й процентили), неспособность процветать была диагностирована (диаграммы роста CDC / WHO 2000). В возрасте 6 месяцев пациентка перенесла трансплантацию гематологических стволовых клеток у здоровой 5-летней сестры с соответствием HLA 10/10 в качестве донора. После кондиционирования, состоящего из тиотепа, treosulfan и флударабина, он получил CD34 + клетки (24,7 × 10 6 клеток / кг веса тела). Циклоспорин А, метотрексат и антитимоцитарный глобулин были даны в качестве профилактики для трансплантат против хозяина (РТПХ) (рис (рис.3).3). Пациенты с синдромом ICF часто обнаруживают остаточную функцию Т-клеток, что может привести к отказу или отторжению трансплантата, и поэтому они подвергаются снижению интенсивности ( 3 ). Учитывая хороший статус работы нашего пациента, вводили миелоаблативное кондиционирование с пониженной токсичностью для достижения стабильного приживления лимфогематопоэза с низким риском токсичности органов ( 12 ).
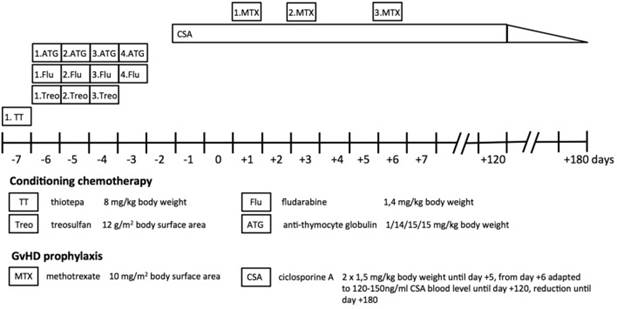
Рисунок 3
Режим лечения. Миелоаблативная кондиционирующая химиотерапия перед трансплантацией состояла из тиотепа, флударабина, треосульфана и антитимоцитарного глобулина. Циклоспорин А и метотрексат назначались как иммуносупрессивная профилактика заболевания трансплантата против хозяина (GvHD) после трансплантации.
Он страдал от острых химиотерапевтических реакций в слизистой оболочке (токсичность 4-го класса ВОЗ) и на коже (токсичность 1-го класса ВОЗ), тошнота и рвота (класс 2-й степени ВОЗ) и почечная недостаточность (токсичность токсичности 2-го класса ВОЗ). Со временем он полностью оправился от всех побочных эффектов. Приживление лейкоцитов и нейтрофилов было достигнуто в день +21 и в день +22, соответственно. Анализ химеризма крови показал аутологичную часть максимум в 0,5%, тогда как химеризм костного мозга был полностью от донора. Без каких-либо инфекций или признаков ГВД, он был выписан из больницы в день +47. Он продолжал получать циклоспорин А до дня +120, который постепенно уменьшался в отсутствие каких-либо признаков ГВД. Через три месяца после трансплантации, он снова был госпитализирован с инфекцией RSV, которую успешно лечили с помощью Рибавирина. Через четыре месяца после трансплантации он снова был госпитализирован из-за диареи, обструктивного бронхита и лихорадки. Сначала было начато лечение антибиотиками и прекращено, когда в горле обнаружены грипп, RSV и вирус короны. Без специфической антивирусной терапии он полностью выздоровел. Через пять месяцев после трансплантации он представил диарею, субфебрильную температуру и частично компенсированный метаболический ацидоз. Ротавирус обнаруживался в стуле. IV-флюидная терапия в качестве амбулаторного пациента была достаточной для нормализации газов крови, и симптомы отступали спонтанно. Через шесть месяцев после трансплантации аденовирус обнаруживался в крови, стуле и горле без каких-либо клинических симптомов.
Его дифференциальные показатели крови постоянно увеличивается и в норме через 6 месяцев после трансплантации (таблица (Таблица1).1 ). Химеризм в костном мозге показал общую аутологичную часть 1-5%, с 1-5% в фракциях CD3 + и CD33 +, а также 0-1% в подмножестве CD34 +. Химеризм в крови был с 2,9% аутологичными клетками, стабильными в течение последних 4 месяцев.
В возрасте 1 года его неврологическое развитие коррелировало примерно с 7-месячным ребенком. Его масса тела составляла 8590 г (9-й процентиль) и длина 75 см (18-й процентиль) (диаграммы роста CDC / WHO 2000).
Идти к:
Обсуждение
Мы сообщаем об успешном аллогенном HSCT 1-летнего мальчика с синдромом ICF1, несущим гомозиготную мутацию в гене DNMT3B после получения клеток костного мозга из 10/10 HLA-подобранной клинически здоровой сестры. В течение 1 года наблюдения мы наблюдали полное восстановление иммунитета без острых или краткосрочных осложнений, связанных с трансплантацией.
Иммунодефицит, центромерная нестабильность и синдром лицевой аномалии - это редкое и тяжелое заболевание иммунодефицита. Наш пациент является пятым ребенком родственных родителей и прошел ХСКТ в раннем возрасте 6 месяцев. Первая дочь тех же родителей умерла в возрасте 5 месяцев в 2009 году в Марокко из-за респираторной недостаточности неизвестного происхождения. Поскольку дальнейшая диагностика не проводилась посмертно , мы можем только предположить, что одна и та же гомозиготная мутация в гене DNMT3B могла быть причиной ее ранней смерти. Хотя гендерная предвзятость для синдрома ICF2, вызванная мутацией в ZBTB24ген наблюдался недавно, синдром ICF1 влияет на обоих полов, что делает еще более правдоподобным, что умершая дочь имела тот же генетический дефект, что и наш пациент ( 13 ).
В этой семейной истории подчеркивается тот факт, что пациенты ICF имеют очень плохие клинические результаты, если их не лечат рано и адекватно. В случае сообщений с 1990-х годов только пациенты на Ig-терапии выжили до детства, а другие умерли в течение первого года жизни от оппортунистических инфекций ( 9 ), как описано в девяти случаях МКФ во Франции. В последнее время Gennery et al. опубликовал успешное лечение иммунодефицита HSCT у трех пациентов ICF, получивших HSCT в возрасте 18 месяцев, 2 и 4 года ( 3). Здесь мы сообщаем еще одно дело об успешном HSCT при синдроме ICF, которое получило HSCT в возрасте 6 месяцев. Сам HSCT связан с высоким риском смертности из-за краткосрочных оппортунистических инфекций и ГВД. Длительные побочные эффекты включают химиотерапевтическую токсичность, такую как дисфункция печени или почек, замедление роста, бесплодие и вторичные злокачественные опухоли ( 14 ). Пациенты, которые ранее не получали химиотерапевтическое лечение, обычно имеют лучший клинический результат и могут успешно трансплантироваться с небольшим острым побочным эффектом, как это наблюдается у пациентов с гемоглобинопатиями или серповидноклеточной анемией ( 15 , 16 ).
Поскольку тяжелые инфекции значительно ухудшают результаты пациентов после HSCT, мы стремимся как можно раньше выполнять ХСКТ при синдроме МКФ в период без инфекции. Синдром ICF неизменно ассоциируется с низким или отсутствующим уровнем иммуноглобулинов, учитывая дефектную дифференцировку периферических терминальных В-клеток. Действительно, к этому можно относиться с заменой IgG на всю жизнь. Тем не менее, у ряда из этих пациентов также проявляется функциональный иммунодефицит Т-клеток, о чем свидетельствует наш случай пневмонией ПЖ . Кроме того, недавние исследования, проведенные Rechavi et al. и Weemaes et al. подчеркнуть тот факт, что дефект пролиферации Т-клеток развивается со временем ( 4 , 17). Таким образом, пациенты с синдромом МКФ страдают от многосистемного заболевания, приводящего к длительной заболеваемости и значительной смертности. Это обычно связано с серьезными инфекциями (т. Е. В тракте GI), связанными с задержкой роста и ограниченной продолжительностью жизни. В частности, на поздней стадии заболевания HSCT ассоциируется с более высоким риском смертности из-за ранее существовавших инфекций. Таким образом, аллогенный HSCT является привлекательным терапевтическим подходом у пациентов с признаками Т-клеточного иммунодефицита без тяжелой предшествующей заболеваемости и обычно рекомендуется, когда идентифицируется HLA-подобный донор. Хотя кондиционирующая химиотерапия сопровождается высокой токсичностью, мы считаем, что преимущества HSCT значительно перевешивают курс естественных болезней в ICF. HSCT заменяет все кроветворные клетки, несущиеDNMT3B и, таким образом, может вылечить иммунодефицит у пациентов с МКФ. Weemaes et al. и Hagleitner et al. исследовали клинические особенности пациентов ICF1, которые могут помочь предсказать клинические результаты после HSCT ( 4 , 7 ). Периодические инфекции и более высокий риск развития гемато-онкологических злокачественных опухолей устраняются у нашего пациента с HSCT, в то время как отсроченное неврологическое развитие или атрофия коры головного мозга с судорогами являются признаками, которые остаются ( 4 , 7). Во время иммунодепрессивного лечения и до восстановления адаптивной иммунной системы у нашего пациента были рецидивирующие респираторные и желудочно-кишечные инфекции. Это может быть связано с посттрансплантационной иммуносупрессией и, как ожидается, со временем улучшится. В то время как неспособность процветать может быть связана с высокой частотой инфекций, мы ожидаем, что наш пациент наверстает увеличение веса в ближайшие месяцы.
Метилирование ДНК представляет собой динамический процесс, который эпигенетически регулирует экспрессию гена во время развития и другие процессы, такие как старение, канцерогенез или клеточная дифференциация. Было описано, что мутация DNMT3B приводит к изменению эпигенетических модификаций генов, регулирующих развитие, нейрогенез и иммунную функцию ( 18). Поскольку ДНК-метилтрансфераза представляет собой внутриклеточный фермент, который не секретируется и не поглощается окружающей тканью, HSCT не может компенсировать дефицит фермента во всех других типах клеток организма. Недавнее исследование показывает рядом с гипометилированием перицентромерных областей хромосом 1, 9 и 16, глобальная потеря метилирования у фибробластов пациентов ICF и линий лимфобластоидных клеток приводит к значительно усиленной экспрессии генов дифференцированных метилированных позиций ( 19). Хотя было идентифицировано 181 таких регионов, остается неясным, как и в какой степени на данную ткань влияет гипометилирование и в какой степени эта картина изменяется во время развития. На все эти вопросы нужно ответить, прежде чем мы сможем перевести молекулярные данные в клинический фенотип. Тем не менее, мы можем предположить, что экспрессивная экспрессия гена может оказать большое влияние на гены, которые имеют высокую эпигенетическую регуляцию, такие как те, которые ответственны за неврологическое развитие или гены зародышевой линии, важные для предотвращения развития рака. Оба из них могут по-прежнему приводить к задержанному неврологическому развитию и более высокому риску злокачественных новообразований у пациентов с МКФ даже после успешного HSCT.
Идти к:
Заключительные замечания
Увидев комбинацию легкой аномалии лица, агаммаглобулинемии в присутствии нормального количества В-клеток и оппортунистических инфекций у пациента, врачи должны рассматривать синдром ICF как дифференциальный диагноз. HSCT является единственным лечебным вариантом иммунного дефекта и должен быть проведен рано, чтобы улучшить выживаемость. Тем не менее, результаты развития и неврологического исхода остаются в значительной степени неизменными, поскольку эпигенетическая дисрегуляция негематопоэтических клеток не может быть исправлена с помощью HSCT.
Тяжелая энтеропатия и гипогаммаглобулинемия, осложняющая рефрактерную микобактериальную туберкулезную комплексную диссеминированную болезнь у ребенка с дефицитом IL-12Rβ1
Абстрактно
Цель
Менделевская восприимчивость к микобактериальному заболеванию является редким клиническим заболеванием, характеризующимся предрасположенностью к инфекционным заболеваниям, вызванным плохо вирулентными микобактериями. Сообщается также о других инфекциях, таких как сальмонеллез и кандидоз. Цель этой статьи - описать молодого мальчика, страдающего различными инфекционными заболеваниями, вызванными комплексом Mycobacterium tuberculosis , Salmonella sp , Klebsiella pneumonie , Citrobacter sp. И Candida sp, осложненным тяжелой энтеропатией и транзиторной гипогаммаглобулинемией.
Методы
Мы проанализировали медицинские записи и провели окрашивание проточной цитометрии популяций лимфоцитов, пролиферацию лимфоцитов в ответ на PHA и продукцию внутриклеточного IFN- γ в T-клетках PHA-бластов у пациента и здоровый контроль. Секвенсирование Sanger использовалось для подтверждения генетических вариантов у пациента и родственников.
Результаты
Генетический анализ выявил биаллельную мутацию в IL12RB1 (C291Y), что привело к полному дефициту IL-12Rβ1. Функциональный анализ продемонстрировал отсутствие внутриклеточного продуцирования IFN-γ в CD3 + Т-лимфоцитах у пациента в ответ на rhIL-12p70.
Выводы
Насколько нам известно, это третий пациент с МСМД из-за дефицита IL-12Rβ1, осложненного энтеропатией и гипогаммаглобулинемией, и первый случай этого заболевания, который будет описан в Колумбии.
Ключевые слова
Микобактерии гипогаммаглобулинемии энтеропатия ИЛ-12Рβ1 дефицит
Менделевская восприимчивость к микобактериальному заболеванию является редким клиническим заболеванием, характеризующимся предрасположенностью к инфекционным заболеваниям, вызванным плохо вирулентными микобактериями. Сообщается также о других инфекциях, таких как сальмонеллез и кандидоз. Цель этой статьи - описать молодого мальчика, страдающего различными инфекционными заболеваниями, вызванными комплексом Mycobacterium tuberculosis , Salmonella sp , Klebsiella pneumonie , Citrobacter sp. И Candida sp, осложненным тяжелой энтеропатией и транзиторной гипогаммаглобулинемией.
Методы
Мы проанализировали медицинские записи и провели окрашивание проточной цитометрии популяций лимфоцитов, пролиферацию лимфоцитов в ответ на PHA и продукцию внутриклеточного IFN- γ в T-клетках PHA-бластов у пациента и здоровый контроль. Секвенсирование Sanger использовалось для подтверждения генетических вариантов у пациента и родственников.
Результаты
Генетический анализ выявил биаллельную мутацию в IL12RB1 (C291Y), что привело к полному дефициту IL-12Rβ1. Функциональный анализ продемонстрировал отсутствие внутриклеточного продуцирования IFN-γ в CD3 + Т-лимфоцитах у пациента в ответ на rhIL-12p70.
Выводы
Насколько нам известно, это третий пациент с МСМД из-за дефицита IL-12Rβ1, осложненного энтеропатией и гипогаммаглобулинемией, и первый случай этого заболевания, который будет описан в Колумбии.
Ключевые слова
Микобактерии гипогаммаглобулинемии энтеропатия ИЛ-12Рβ1 дефицит
Андрес Аугусто Ариас и Карлос М. Перес-Велес, Хасинта Бустаманте и Хосе Луис Франко внесли одинаковый вклад в эту работу
Электронный дополнительный материал
Онлайн-версия этой статьи ( https://doi.org/10.1007/s10875-017-0435-1 ) содержит дополнительный материал, доступный для авторизованных пользователей.
Введение
Менделевская восприимчивость к микобактериальным заболеваниям (MSMD, MIM 209950) является редким унаследованным состоянием, характеризующимся избирательной предрасположенностью к плохо вирулентным микобактериям, таким как штамм Mycobacterium bovis BCG и нетуберкулезные микобактерии (NTM). Пациенты, затронутые этой первичной инфекцией иммунодефицита (ПИД), также уязвимы к более вирулентным видам комплекса Mycobacterium tuberculosis [ 1 ]. Десять генов, вызывающих MSMD, включая восемь аутосомных ( IFNGR1 , IFNGR2 , STAT1 , IL12B , IL12RB1 , ISG15 , TYK2 и IRF8 ) и два X-связанных (NEMO и CYBB ) были обнаружены до сих пор. Высокий уровень аллельной гетерогенности уже привел к определению 20 различных расстройств, все из которых связаны с иммунологическим иммунитетом IFN-γ [ 1 ]. Наиболее распространенная генетическая этимология МСМД является результатом аутосомно-рецессивных (AR) мутаций в IL12RB1, кодирующих цепь IL-12Rβ1 рецепторов IL-12 и IL-23 [ 2 ]. Все мутантные аллели являются потерями функции, и лимфоциты пациентов не реагируют на IL-12 и IL-23 и продуцируют низкие уровни IFN-γ [ 2 , 3 , 4]. Клинический фенотип гетерогенен и характеризуется главным образом микобактериальными инфекциями, за которыми следуют сальмонеллез, кандидоз и туберкулез, а реже - инфекции, вызванные Klebsiella pneumoniae , Paracocci-dioides brasiliensis , Coccidioides spp. И Histoplasma spp. [ 2 ]. Здесь мы сообщаем о ребенке с дефицитом IL-12Rβ1, который подавал диссеминированную болезнь M. bovis BCG после вакцинации BCG (BCG-osis), включая хронический энтерит и энтеропатию, а также переходную гипогаммаглобулинемию. Впоследствии он также разработал грамотрицательные бактериальные инфекции (сальмонеллез, клебсиеллез, цитробактериоз) и слизисто-кожный кандидоз.
История болезни
Пациентка - мужчина, родившийся в 2009 году в городе Перейра (Колумбия, Южная Америка) от явно не относящихся к кровным родителям родителей в возрасте 39 недель гестационного возраста. У него был старший брат, который умер в возрасте 5 лет из-за BCG-osis. Пациент весил 3,350 г при рождении и получал вакцину БЦЖ в соответствии с руководящими принципами национальной программы иммунизации Колумбии наряду с другими запланированными вакцинами до 6 месяцев, когда вакцинация была приостановлена по неизвестным причинам. Его первые симптомы начались в возрасте 4 месяцев, когда он развил бессимптомный узел в левой подмышечной области. Мать отвела его к педиатру, который сделал клинический диагноз регионального гнойного лимфаденита после вакцинации БЦЖ («БЦЖ-итис»), но решил следить за его прогрессом без лечения и выписывать его домой и назначать последующие тесты. Через месяц он снова был замечен, но на этот раз конкреция превратилась в абсцесс, и рентгенография грудной клетки не выявила аномалий, а результаты полного анализа крови (CBC) и группы химии крови находились в нормальных пределах. Оральный диклоксациллин назначался без терапевтического ответа, и потом ребенок был госпитализирован и начинался с внутривенного (IV) оксациллина. Диагноз вторичного лимфаденитаM. bovisБЦЖ был рассмотрен, но ребенок оставался афебрильным и хорошо питался; поэтому он был уволен с последующим наблюдением в течение 1 месяца. В возрасте 11 месяцев ребенок снова был госпитализирован, поскольку у него развились стойкие лихорадки, раздражительность и неспособность развиваться, а лимфаденит оставался гнойным, по этой причине снова вводили оксациллин IV. Во время физического обследования отмечались шейные лимфаденопатии и гепатоспленомегалия, а дифференциальный диагноз включал лимфому и диссеминированный туберкулез (ТБ). Торако-абдоминальная контрастная улучшенная компьютерная томография (CECT) выявила множественные лимфаденопатии в области шеи, как подмышечные, так и верхние средостения, которые соответствовали диффузному заболеванию лимфатических узлов, а также асциту в брюшной полости; печень и селезенка были нормальными. Микроскопии серийного мазка разряда из подмышечной впадины и желудочных аспиратов были положительными для кислотоустойчивых бацилл (AFB). В то же время, на основе предполагаемого диссеминированного заболевания из-заM. tuberculosis , антимикобактериальное лечение было начато с пероральным изониазидом (H), рифампицином (R) и пиразинамидом (Z), 6 дней в неделю в течение 6 месяцев, и ребенок был выгружен в ожидании результатов микобактериальных культур. Через месяц культуры гноя из подмышечной впадины и желудочных аспиратов стали положительными для микобактерий, и тест на ниацин был признан положительным, а изоляты были отправлены в Национальный институт здравоохранения Колумбии для тестирования на чувствительность к лекарственным препаратам (Bactec MGIT 960, Becton Dickinson , Franklin Lakes, NJ), демонстрирующие восприимчивость к H, R, Z, стрептомицину (S) и этамбутолу (E).
В возрасте 15 месяцев ребенок был госпитализирован из-за левой подмышечной лимфаденопатии, для которой он подвергся резекции, а гистопатология выявила гранулематозное воспаление с некрозом, обильными гигантскими клетками и гистиоцитами, но AFB не были обнаружены. Однако новые мазки желудочного аспирата были AFB-положительными. Новый торако-абдоминальный CECT выявил снова асцит и диффузное заболевание лимфатических узлов, и, хотя не было признаков или симптомов, указывающих на инфекцию ЦНС, проводилась поясничная пункция, а количество клеток и химия спинномозговой жидкости (CSF) находились в пределах нормы и микроскопия мазка была отрицательной для AFB. Из-за озабоченности по поводу рецидива ранее лечившегося туберкулезного заболевания был рассмотрен туберкулез с множественной лекарственной устойчивостью (МЛУ-ТБ) и добавлен внутримышечный (IM) S (первоначально назначенный на 60 дней, но затем прекращалось через 22 дня), а затем лечение де-эскалации до H плюс R 3 раза в неделю по причинам, не ясным из медицинских записей. Микобактериальные культуры желудочного аспирата и CSF были отрицательными через 2 и 3 месяца, соответственно.
В возрасте от 16 до 19 месяцев ребенок представил два эпизода не-кровавой диареи, рвоту, снижение аппетита, прогрессирующее растяжение брюшной полости и постоянные лихорадки, предположительно вызванные желудочно-кишечными (ГИ) вирусными инфекциями. Они были обработаны жидкостью для дегидратации и пробиотиков с разрешением симптомов. В возрасте 20 месяцев ребенок был госпитализирован для последующего наблюдения, и были получены желудочные аспираты для микроскопии мазка и микобактериальной культуры, и все они были отрицательными. Однако к 21 месяцу диарея стала устойчивой и тяжелой, что привело к обезвоживанию диурией и олигурией наряду с отек лица. Болезнь кишечника и почек была вызвана, и он снова был госпитализирован. Гипоальбуминемия была зарегистрирована (2,96 г / дл, ссылка 3.8-5,4 г / дл), и, кроме того, сывороточный креатинин был нормальным, а образцы мочи выявляли пирурию, но не протеинурию, а мазки ПФ и микобактериальные культуры были отрицательными. Ультразвук с высоким разрешением почечной недостаточности выявил нормальные почки и асцит, экскреторная урография показала только воспаление сбора чашечек, а почечная проверка димеркаптоянтарной кислоты (DMSA) не документировала активную инфекцию. К тому времени антимикобактериальное лечение состояло только из H плюс R 3 раза в неделю. Тем не менее, пациент продолжал ухудшаться, что приводило к подозрению в лежащем в основе ПИД-инфекции, и ребенка направляли на диагностику туберкулеза, страдающего от рефрактерного туберкулеза, на педиатрического специалиста по туберкулезу в третичном центре в Медельине для дальнейшей оценки в возрасте 22 месяцев ,
После поступления в этот центр на физическом обследовании был выявлен тяжело дегидратированный и недоедающий ребенок (вес-на-рост (W / H) -0,65, рост по возрасту (H / A) -2,06) с шейным, подмышечным, подчелюстным, и паховые лимфаденопатии; сильное брюшное растяжение; и отек нижних конечностей. CBC показал умеренную лейкопению и лимфопению с нормальными моноцитами и тромбоцитами (дополнительная таблица 1 ). Гипопротеинемия и гипоальбуминемии были также отмечены (3,84 г / дл; реф 5 .6-7.5 и 1,47 г / дл; исх 3 .8-5.4, соответственно, Справочная таблица 2). Кроме того, сывороточные АСТ и АЛТ и билирубины находились в нормальных пределах, а серологическая и вирусная нагрузка для теста на ВИЧ и PPD-туберкулин были отрицательными. На этот раз торако-абдоминальный CECT не обнаружил аномалий в сундуке, но в брюшной полости он обнаружил множественные забрюшинные лимфаденопатии и асцит, но без органомегалий. Антимикобактериальное лечение проводилось в течение 2 недель для улучшения бактериологического выхода новой коллекции образцов (кровь, стул, серия аспирата желудка, биопсия подмышечного лимфатического узла и асцитическая жидкость). Биопсия лимфатических узлов выявила обильное некротизирующее гранулематозное хроническое воспаление, которое было положительным для AFB. Тем не менее, микроскопия мазков AFB, микобактериальные культуры и МТК (MTC) ПЦР (Xpert MTB / RIF®, Cepheid, Sunnyvale, CA) всех других образцов были отрицательными. Несколько дней спустя, устный дрозд был замечен, и кандидоз был диагностирован и лечился с полным разрешением. Наконец, антимикобактериальное лечение презумптивного лекарственно-устойчивого ТБ было начато снова с помощью H, R, Z, E плюс этионамида (Eto) и левофлоксацина (L), а также агрессивного энтерального питания и инфузий альбумина, что привело к разрешению асцита и отека нижних конечностей, но не хронической диареи. Ребенка обсуждали с экспертами PID и с сильным подозрением на MSMD, пациент был переведен в возрасте 24 месяцев в реферальный центр PID в Медельине. наряду с агрессивным энтеральным питанием и инфузиями альбумина, что приводит к разрешению асцита и отека нижних конечностей, но не хронической диареи. Ребенка обсуждали с экспертами PID и с сильным подозрением на MSMD, пациент был переведен в возрасте 24 месяцев в реферальный центр PID в Медельине. наряду с агрессивным энтеральным питанием и инфузиями альбумина, что приводит к разрешению асцита и отека нижних конечностей, но не хронической диареи. Ребенка обсуждали с экспертами PID и с сильным подозрением на MSMD, пациент был переведен в возрасте 24 месяцев в реферальный центр PID в Медельине.
В реферальном центре проводилась эндоскопия верхней и нижней желудочно-кишечного тракта (ГИТ) с хроническим неспецифическим воспалением, но без атрофии; окрашивание всех биопсии участков было отрицательным на AFB (рис. 1а ); Кроме того, культуры всех биопсий не изолировали микобактерии. Первая ПЦР для MTC и NTM (Genotype Mycobacteria Direct®, Hain LifeScience GmbH, Германия) от биопсиотической лимфаденопатии была положительной для MTC; однако вторая ПЦР для MTC (Xpert MTB / RIF®), выполненная несколькими неделями позже в одном желудочном аспирационном образце, была отрицательной. CBC оставался в основном в нормальных пределах, но с легкой до умеренной лимфопении во время пребывания в больнице (дополнительная таблица 1); белки сыворотки, измеренные несколько раз во время этой госпитализации, выявили общие и белковые общие белки и альбумин (дополнительная таблица 2 ) и тяжелая гипогаммаглобулинемия (0,17 г / дл, 0,6-1,6 г / дл).
Рисунок 1
эндоскопии и Н & Е окрашивание биопсии двенадцатиперстной кишки показывает умеренное хроническое воспаление с мононуклеарными инфильтратами и не атрофии (× 40). b Покрытие проточной цитометрии клеток EBV-B с анти-CD212 или изотипом при здоровом контроле, пациенте и отрицательном контроле (EBV-B-клетки у пациента с дефицитом IL-12Rβ1). c Т-клеточные PHA-бласты от здорового контроля, пациент и его мать либо не стимулировались (NS), либо стимулировали в течение 16 ч 20 нг / мл rhIL-12p70. Внутриклеточное продуцирование IFN- γ оценивали с помощью проточной цитометрии путем окрашивания живых клеток (отрицательная популяция Aqua) и исключения агрегатов с помощью FSC-H и FSC-A и стробирования на CD3 + T-лимфоцитах. dЭлектроферограмма показывает мутацию C291Y у пациента и родителей. eРодословная с указанием индекса, умершего брата и обоих родителей; каждое поколение имеет римскую цифру (I-II), стрелка указывает на пробанду. Никакая ДНК не была доступна от брата для мутационного тестирования («E?»)
После этого измеряли уровни Ig в сыворотке крови, выявляя низкий IgG (246 мг / дл, 749-2682 мг / дл), нормальный IgM и IgA и слегка повышенный IgE (178,3 МЕ / мл, 0,1-1,8,8 МЕ / мл соответственно) (Дополнительный рисунок 1 ). Фенотипирование T, B и NK-клеток проточной цитометрией в настоящее время невозможно. Для дальнейшего изучения генетической причины его болезни трансформированные В-клетки вируса Эпштейна-Барра (EBV) окрашивали флуоресцентно меченным анти-IL-12Rβ1 mAb, выявляя полное отсутствие экспрессии белка с помощью проточной цитометрии ( фиг.1b); аналогичные результаты были получены из Т-клеток периферической крови (PB) (не показаны). Кроме того, стимулированные rhIL-12p70 PB-лимфоциты не показали внутриклеточного продуцирования IFN-γ в CD3 + T-лимфоцитах у пациента по сравнению со здоровым контролем и гетерозиготной матерью по проточной цитометрии ( рис.1c ). Анализ последовательности геномной ДНК всех кодирующих экзонов IL12RB1выявил гомозиготную миссенсусную мутацию в экзоне 9 (c.872G> A), который заменил цистеин в положении 291 тирозином (p.C291Y) (3), подтвердив диагноз дефицита IL-12Rβ1 ( Фиг.1d ); оба родителя были гетерозиготными для одной и той же мутации, но биологический материал не был доступен для проверки умершего брата (рис. 1е). При силиковой оценке мутации p.C291Y с оценкой Полифен-2, SIFT и CADD (максимум 25,3) указывали, что этот вариант повреждает.
Учитывая неопределенность в отношении того, были ли обнаружены обнаруженные виды MTC M. tuberculosis или M. bovis BCG, пиразинамид (который не лечил M. bovisBCG) был заменен левофлоксацином (который будет лечить оба вида), а H, R и E были продолжение. Рекомбинантный человеческий (rh) IFN-γ добавляли в возрасте 28 месяцев в дозе 200 мкг / м 2, 3 раза в неделю. Уровни IgG сыворотки измеряли серийно в течение 2 месяцев и оставались низкими; поэтому внутривенный иммуноглобулин (IVIG) был начат в возрасте 27 месяцев в дозе 600 мг / кг первоначально каждые 15 дней в течение 15 месяцев, а затем продолжался ежемесячно в течение еще 10 месяцев, а затем 11 месяцев подкожного иммуноглобулина ( SCIG). Кроме того, добавление энтерального питания с кормлением труб продолжалось до возраста 60 месяцев. Все лечение привело к значительному улучшению его общего состояния с разрешением диареи и полным восстановлением уровней IgG в сыворотке (см. Фиг.1 ); тем не менее уровни сывороточного альбумина оставались низкими до 72-месячного возраста, когда они стали нормальными (дополнительная таблица 2). Распространенный туберкулез лечился в течение 12 месяцев, а затем 24 месяца вторичной профилактики, а SCIG продолжали в течение 36 месяцев. CBC нормализуется, но лимфопения от легкой до умеренной сохраняется через 24 месяца (дополнительная таблица 1 ); CBC колебался со временем, а лимфопения сохранялась вместе с легкой нейтропенией. Недавняя фенотипизация PBL по-прежнему проявляла легкую лейкопению с нормальными Т-лимфоцитами, но слегка низкими суммарными CD3 + Т-клетками и CD3 + CD8 + Т-клетками; однако, лимфопролиферация до фитогемаглутинина (PHA) была нормальной (дополнительные таблицы 3 и 4 ).
У пациента были другие тяжелые инфекции: Salmonella spp. гастроэнтерит в возрасте 3 лет, лихорадочная нейтропения с бактериемией Proteus mirabilis в возрасте 42 месяцев, а через 46 месяцев - бактериальный Citrobacter freundii абсцесс глубокой шеи в подчелюстной области. Для этих инфекций он получил IV антибиотики с хорошим ответом. Через 59 месяцев у него развилась тяжелая диарея, связанная с асцитом, цервикальными лимфаденовыми путями и лейкопенией с лимфопенией и гипоальбуминемией (1,2 г / дл); однако, поскольку он все еще принимал SCIG, его сывороточный IgG был нормальным (дополнительная таблица 3). На этот раз эзофагогастродуоденоскопия выявила эзофагит с белыми бляшками в соответствии с кандидозом, который успешно лечился флуконазолом. Биопсии ЖКТ выявили неспецифическое воспаление. Микобактериальные культуры из крови и биопсии GIT были отрицательными, а также MTC PCR (Xpert MTB / RIF®) биопсии GIT. Однако культуры крови были положительными для штамма штамма K. pneumoniae с расширенным спектром бета-лактамазы (ESBL), который лечился IV антибиотиками с разрешением инфекции. Он также получал альбумин, сульфат цинка и низкую диету FODMAP (ферментируемая, олиго-, ди-, моносахариды и полиолы) и полностью восстанавливался. В настоящее время он по-прежнему находится на rhIFN-γ и профилактическом триметоприм-сульфаметоксазоле (TMP-SMX) три раза в неделю.
Обсуждение
Мы описываем первого колумбийского ребенка с МСМД из-за гомозиготной мутации в IL12Rβ1 и поражены различными инвазивными инфекционными заболеваниями, связанными с дефицитом IL-12Rβ1, включая микобактериальный (MTC), бактериальный (не тифоидный Salmonella , K. pneumoniae , C. freundii ) , и грибковые ( CandidaSPP.). Мы не могли твердо установить виды МТК, поскольку изолят во время первоначального микробиологического диагноза не был представлен для дальнейшего видообразования, а микобактериальное тестирование различных образцов, собранных во время рецидивов распространенного заболевания МТК, было отрицательным по культуре, несмотря на то, положительный с помощью микроскопии мазка и ПЦР MTC, предполагая, что бациллы были нежизнеспособными, возможно, из-за недавней антимикобактериальной терапии или обработки образцов. Вначале мы предположили, что этот вид был M. bovis BCG с учетом рецидива левого подмышечного лимфаденита в раннем возрасте у вакцинированного BCG ребенка, связанного с вовлечением удаленного органа (т. Е. GIT) и отсутствием легочного поражения в установлении PID, хорошо распознанного для предрасположенности к M. bovisРаспространенная БЦЖ болезнь. Однако, тот факт , что первоначальный МТК изолят был и ниацин-позитивный, так и пиразинамид-восприимчивый, маловероятен , чтобы были М. Bovis БЦЖ, так как подавляющее большинство изолят ниацина-отрицательное и пиразинамид устойчивости [ 5 , 6 ] , С другой стороны, хорошо известно, что заболевание M. tuberculosis, связанное с дефицитом IL-12Rβ1, не очень редко и что клинический синдром диссеминированного заболевания может возникать у детей, которые в противном случае невосприимчивы к слабо вирулентным микобактериям, таким как M. bovis BCG и нетуберкулезным микобактериям [ 7 , 8]. Тем не менее, наиболее распространенной инфекцией, обнаруженной у пациентов с дефицитом IL-12Rβ1, по-прежнему остается заболевание M. bovis BCG, распространенное через несколько месяцев после вакцинации [ 1 ]. Такие события, предположительно связанные с вакцинацией или иммунизацией, включая осложнения, связанные с вакцинацией БЦЖ (такие как БЦЖ-итид и БЦЖ-озис), должны считаться серьезными и быстро сообщаться через эффективные системы эпидемиологического надзора, чтобы обеспечить своевременную диагностику пациентов, страдающих МСМД и другие ПИД, которые приводят к восприимчивости к этим микроорганизмам [ 9 , 10 ].
Значительным нахождением у этого пациента было развитие тяжелой гипопротеинемии и гипогаммаглобулинемии, скорее всего, в контексте хронической диареи и тяжелого недоедания, поскольку печень и почечная дисфункция никогда не были продемонстрированы. Кроме того, мы считаем, что воспаление GIT (как показано на рисунке 1a ) могло бы способствовать потере белков, что указывает на белково-потерю энтеропатии (PLE); однако для подтверждения этого не было никаких конкретных тестов на биомаркер. Насколько нам известно, было только два случая, сообщающих об ассоциации дефицита IL-12Rβ1 с энтеропатией, истощающей белок или тяжелой гастроэнтеропатией. Первым был 4-летний мексиканский мальчик, страдающий слизисто-кожным кандидозом, и M. bovisРаспространенная БЦЖ болезнь, которая умерла, несмотря на множественные антимикобактериальные методы лечения. Однако у этого пациента не было отчета о биопсии GIT, и PLE не было документировано; кроме того, уровни Ig сыворотки были нормальными [ 11 ]. Второй пациент был 49-летним кавказским мужчиной с Фарерских островов (Дания), который страдал тяжелой гастроэнтеропатией и потерей веса в течение 2-летнего периода [ 12 ]. Интересно, что гастродуоденоскопия выявила дуоденит, но, опять же, PLE не была продемонстрирована. Микроскопия биопсий GIT выявила AFB, но микобактериальные культуры были отрицательными; тем не менее, амплификация и секвенирование сегмента гипервариабельной области 16S рРНК-ДНК выявили высокую гомологию с последовательностями ДНК микобактериальной РНК, предполагая Mycobacterium branderi, У него также была гипоальбуминемия, но сывороточный IgG и IgM были нормальными, тогда как IgA не определялся.
У пациентов с дефицитом IL-12Rβ1 обычно наблюдается относительно мягкое клиническое заболевание и хороший прогноз [ 3 , 13 ]. В большинстве случаев их можно лечить с помощью пролонгированной антимикобактериальной терапии (2-3 года), rhIFN-γ и антимикробной терапии по мере необходимости при сопутствующих инфекциях, а также для профилактики, если инфекции повторяются. rhIFN-γ эффективен при традиционной терапии для случаев рефрактерного микобактериального диссеминированного заболевания, такого как наш пациент [ 14 , 15 , 16 ]. У него положительный ответ на rhIFN-γ, хотя он показал умеренную лейкопению (нейтропения и лимфопения), которые могут быть отчасти объяснены этой терапией [ 17], а также профилактическому TMP-SMX; по этой причине мы регулярно наблюдаем за пациентом, чтобы предотвратить возможные осложнения. Хирургическое удаление брыжеечных лимфатических узлов часто указывается для локализованных инфекций с плохим ответом на антимикобактериальную терапию [ 1 ].
Мы подчеркиваем, что все пациенты, у которых развиваются осложнения от MTC (независимо от того, являются ли M. tuberculosis , M. bovis BCG или другие MTC), должны оцениваться для ПИД и управляться соответственно при помощи специалистов в области детского туберкулеза и ПИД. Наконец, вполне вероятно, что оба родителя разделяют некоторую степень родства на основе их общего происхождения из небольшого и закрытого сообщества [ 18 , 19 ], что указывает на необходимость рассматривать генетическое заболевание в таких закрытых сообществах и расширять тестирование для родственников.
Этот случай подчеркивает тот факт, что виды MTC, отличные от M. bovis BCG, могут имитировать клиническое проявление подмышечного лимфаденита, ипсилатетра, к месту вакцинации. MTC, за исключением M. bovis BCG, следует подозревать, когда подмышечный лимфаденит выражен на двусторонней основе, а когда виды MTC являются ниациноположительными и пиразинамид-восприимчивыми. Иммунные тесты на туберкулезную инфекцию, такие как анализы на высвобождение IFN-γ (QuantiFERON-TB® (Qiagen, Hilden, Germany) или T.SPOT-TB® (Immunotec, Oxford, UK)) полезны - когда положительные - в том, что они специфичны для вирулентных видов MTC, таких как M. tuberculosis и M. bovis , и не имеют перекрестной реактивности с M. bovisBCG. Однако их чувствительность значительно снижается у пациентов с дефектами в их путях IFN-γ, в том числе с МСМД [ 20]. В глобальном контексте этот случай имеет значение, поскольку клинический сценарий пациента с заболеванием туберкулеза, который невосприимчив к соответствующему антимикобактериальному лечению, или который имеет удовлетворительный терапевтический ответ, но впоследствии имеет рецидив заболевания ТБ, не является чем-то необычным. Когда тестирование на чувствительность к лекарственным препаратам невозможно (что имеет место у подавляющего большинства детей с туберкулезом, которые являются paucibacillary), следует учитывать первичный лекарственно-устойчивый туберкулез, особенно в странах, где МЛУ-ТБ является эндемическим. Однако, когда адекватное лечение лекарственно-восприимчивого рассеянного туберкулеза завершено, а реинфекция считается невероятной, дифференциальный диагноз рецидивирующего заболевания туберкулезом, который распространяется, должен включать МСМД, но недооценивается в литературе по туберкулезу [ 1 ].
Вывод
Мы сообщаем о ребенке, у которого гомозиготная мутация в IL-12Rβ1, связанная с затяжным рефрактерным диссеминированным заболеванием MTC (не указанная, но, вероятно, не M. bovis BCG) была осложнена энтеропатией и гипогаммаглобулинемией. Таким образом, у ВИЧ-неинфицированных детей с инфекционным заболеванием из-за слабо вирулентных микобактерий (например, Mycobacterium bovis BCG, других видов MTC или NTM), интрамакрофагических бактерий (например, сальмонеллы ) и / или грибов (например, Candida), связанный с тем, что он является серьезным (диссеминированным или инвазивным), рецидивирующим или невосприимчивым к лечению, следует учитывать врожденную ошибку иммунитета IFN-γ, такую как IL-12Rβ1, и соответствующие осложнения, такие как энтеропатия и гипогамма-глобулинемия, должны быть простыми признаются, если они присутствуют и управляются надлежащим образом. Эти результаты демонстрируют важность разработки недорогих молекулярных диагностических инструментов, эффективных образовательных программ, доступных лечебных центров и генетического консультирования для пострадавших семей и поощрения дальнейших исследований молекулярной основы МСМД у пациентов [ 19 , 21 ].
Источник: https://www.ncbi.nlm.nih.gov/pubmed/28865061
Дата добавления: 2018-11-24; просмотров: 246; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!