Микроцитогенетические синдромы
В связи с развитием и внедрением в клиническую цитогенетику высокоразрешающих молекулярно-генетических и молекулярно-цитогенетических методов диагностики хромосомных болезней за последнее время удалось выделить особую группу синдромов, обусловленных микроперестройками некоторых хромосом. Разработка и внедрение высокоразрешающих методов в цитогенетику человека не в последнюю очередь были связаны с тем, что возникли затруднения при диагностике хромосомных аномалий, вызванных микроперестройками, которые невозможно было обнаружить с помощью классических рутинных методов окраски хромосом. Достаточно часто такие микроделеции и микродупликации хромосом, вызывающие умственную отсталость и врожденные пороки развития, относились к менделирующим точечным мутациям, наследуемым по аутосомно-доминантному типу. По материалам пре- и постнатальной цитогенетической диагностики частота встречаемости аномальных минихромосом в кариотипе человека колеблется от 1 до 2 случаев на 1000. С помощью молекулярно-цитогенетических методов наконец-то удалось установить истинные причины группы заболеваний с нетрадиционным типом наследования. Частота их встречаемости колеблется от 1:50 000 до 1:100 000 новорожденных.
В основе болезней с нетрадиционным типом наследования могут лежать феномены однородительской дисомии и геномного импринтинга. Следует напомнить, что под явлением однородительской дисомии понимается наличие двух гомологичных хромосом (или хромосомных сегментов) одного из родителей (матери или отца); под геномным импринтингом подразумевают различную экспрессию аллелей в зависимости от их родительского происхождения. Различная активность отцовских или материнских генных локусов может оказывать свое влияние на степень развития плаценты, вес плода, развитие и функционирование других органов и систем. В настоящее время для некоторых хромосом (7, 11, 15) геномный импринтинг твердо установлен для других (2, 3, 6, 14 и 20) предполагается. Геномный импринтинг и однородительская диссомия обусловливают ряд заболеваний человека. К таким заболеваниям относятся синдромы Прадера-Вилли, Ангельмана, Лангера-Гидеона, Беквита-Видемана и др.
|
|
|
Клиническая картина этих заболеваний очень вариабельна. Это может зависеть от протяженности хромосомной микроаномалии, качественного состава вовлеченных генов, от материнского или отцовского происхождения перестройки и т.д. Анализ возникновения микроперестройки и причастность ее к той или иной хромосоме играет очень важную роль при планировании деторождения в семье. Возникать такие аномалии могут на любом этапе гаметогенеза, в том числе и после завершения половой клеткой обоих мейотических делений; другой причиной возникновения частичных анеуплоидий могут стать родительские сбалансированные перестройки (инверсии, транслокации).
|
|
|
Синдром Прадера-Вилли
Впервые синдром Прадера-Вилли был описан в 1956 году. Причиной возникновения этого синдрома является потеря функции хромосомных участков, расположенных в проксимальной части длинного плеча хромосомы 15 (15q11-13). Делеция имеет отцовское происхождение и наблюдается у 70% больных, у 5% заболевание связано с перестройкой хромосомы 15. В большинство случаев заболевание возникает de novo, в 25% случаев синдром возникает в результате однородительской дисомии. У некоторых больных хромосомную аномалию не удается идентифицировать, но у них наблюдается характерная клиническая картина синдрома Прадера-Вилли.

4.5.9
Основными клиническими признаками являются отставание умственного развития, неадекватное поведение, задержка физического развития, низкорослость, гипотония. Одни клинические признаки при этом заболевании можно наблюдать до 3 летнего возраста (мышечная гипотония, малый вес и трудности вскармливания), другие начинают преобладать после 6 месячного возраста (ожирение, усиление аппетита, нарастание умственной отсталости, отставание в росте). Наряду с диспластическими признаками (опущенные углы рта, высокое небо, гипертелоризм, эпикант, маленькие стопы и кисти, миндалевидный разрез глаз, аномалии дерматоглифика) выявляется гипогонадизм, обусловленный низким уровнем половых гормонов, гипопигментация (у75% больных). Следует отметить, что синдром Прадера-Вилли характеризуется широким клиническим полиморфизмом, поэтому необходимо проводить дифференциальную диагностику с синдромами Коэна, Опица-Фриаса, Барде-Бидля.
|
|
|
Продолжительность жизни составляет 25 – 30 лет.
Диагностика заболевания осуществляется с помощью ДНК-анализа или методом FISH. Риск для сибсов пробанда – около 1%.
Синдром Ангельмана
Если для возникновения синдрома Прадера-Вилли основной причиной являлась делеция проксимальной части длинного плеча хромосомы 15 отцовского происхождения, то аналогичная потеря той же части длинного плеча хромосомы 15, но только материнского происхождения обусловливает развитие другой патологии – синдрома Ангельмана. При этом заболевании развивается совсем другая клиническая картина. Для синдрома Ангельмана характерно: выраженная олигофрения, задержка речи, гиперактивное поведение, судороги, большая нижняя челюсть, макростомия, гипопигментация (у 40% больных). Они поздно начинают ходить, для них характерна походка с широко расставленными ногами, локтевые суставы согнуты; отмечается насильственный немотивированный смех, имеются выраженные расстройства координации движений. 
|
|
|
4.5.10
Дифференциальную диагностику следует проводить с синдромами Петерса-Пласа, Ретта и с тригоноцефалией Опица.
Частота синдрома в популяции составляет 1:20 000.
Примерно 20 – 30% больных не имеют делеции проксимальной части длинного плеча хромосомы 15; у незначительного числа больных причиной является однородительская дисомия. Диагностика синдрома осуществляется теми же методами, что и при синдроме Прадера-Вилли, т.е. проводится ДНК-анализ и метод FISH. С помощью этих методов можно установить этиологию около 90% случаев заболевания. Риск для сибсов пробанда не известен.
Синдром Видемана-Беквита.
Не менее интересным и познавательным в плане феномена геномного импринтинга является терминальный район другой хромосомы – район короткого плеча хромосомы 11, структурные и функциональные аномалии которого обусловливают широко распространенный синдром Видемана-Беквита, нефробластому, некоторые опухоли детского возраста.
Синдром Видемана-Беквита достаточно распространенное заболевание, встречающееся с частотой 1:10 000 – 1:12 000 новорожденных.
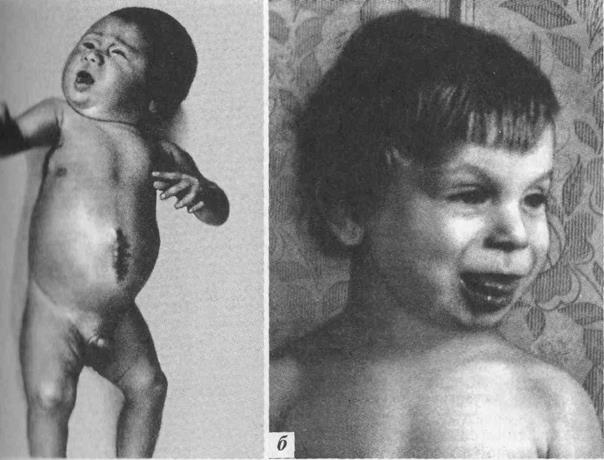
Характерными клиническими признаками этого синдрома являются: гигантизм, пупочная грыжа, макроглоссия, гипоплазия верхней челюсти, прогнатизм, долихоцефалия и другие диспластические стигмы; при рождении отмечается гипогликемия; у больных отмечается повышенная предрасположенность к возникновению опухолей.
4.5.11
Первоначально предпологалось, что синдром Видемана-Беквита наследуется по аутосомно-доминантному и аутосомно-рецессивному типу наследования. Не исключалась возможность и мультифакториального наследования. Но в настоящее время общепринято считать, что синдром наследуется аутосомно-доминантно с неполной пенетратностью и вариабельной экспрессивностью, причем при наследовании большую роль играет феномен геномного импринтинга (в некоторых семьях клинические проявления отмечаются в 15%). Интерес же цитогенетиков был связан с тем, что при синдроме Видемана-Беквита с помощью высокоразрешающих молекулярных методов удалось установить частичную трисомию дистального участка короткого плеча 11 хромосомы отцовского происхождения. Кроме того, в 20% случаев причиной синдрома Видемана-Беквита является однородительская дисомия, а также в некоторых семьях описаны сбалансированные транслокации между 11 и 22 хромосомами, которые вызывают аналогичный синдром. В общей сложности структурные аномалии короткого плеча 11 хромосомы встречаются в 2% и могут быть унаследованы как от матери, так и от отца.
При определении риска у потомства в семьях с синдромом Видемана-Беквита необходимо проводить тщательное молекулярно-генетическое и молекулярно-цитогенетическое обследование.
Лечение хромосомных болезней
Лечение хромосомной патологии в основном симптоматическое. Цель такой терапии заключается в том, чтобы скорректировать такие фенотипические проявления, как умственная отсталость, замедленный рост, недостаточная феминизация или маскулинизация, недоразвитие гонад, устранить или исправить различные костные дефекты ит.д. Для этого широко используют различные виды терапии, в том числе анаболические гормоны, андрогены и эстрогены, гормоны гипофиза и щитовидной железы, различные витамины и общеукрепляющие средства. Очень широко применяется хирургическое симптоматическое лечение: удаление катаракты, лишнего (шестого) пальца на ноге или руке, пластические операции при незаращении верхней губы и/или неба, устранение стеноза привратника и врожденных пороков сердца, удаление различных опухолей и т.д. Перечисленные дефекты часто сопровождают трисомии по хромосомам 13, 18 и 21, триплоидию, синдромы 4р- и 5р- и иные хромосомные аномалии. Из других видов симптоматической терапии следует отметить климатотерапию, бальнеолечение, разные виды электротерапии, теплолечение, рентгенорадиологическое облучение.
Несмотря на широкое разнообразие симптоматической терапии, применяемой для лечения хромосомных болезней, они до сих пор неизлечимы. Учитывая этот факторы, в настоящее время основное внимание уделяется предупреждению рождения детей с хромосомными аномалиями.
Глава 5
МОНОГЕННЫЕ БОЛЕЗНИ
5.1. Общая характеристика моногенной патологии
Моногенные болезни (МБ) – это заболевания, в основе этиологии которых лежит единичная генная мутация. МБ наследуются в соответствии с законами Менделя. В настоящее время описано около 5 000 нозологических единиц МБ. Они выявляются у 3- 6 % новорожденных, а в структуре общей смертности детей до 5 лет на их долю приходится 10-14 %. МБ, гены которых картированы на хромосомах, насчитывают до 900 нозологических единиц. Для примерно 350 болезней выяснен характер генной мутации, установлена природа биохимического дефекта. Для ряда МБ физически картированы на хромосомах конкретные мутантные гены. Индивидуальный и популяционный риск возникновения МБ существенно различаются из-за неравномерного распространения обусловливающих их генов. Принято считать, что МБ, встречающиеся с частотой 1:10 000 и выше – это частовстречающиеся, а с частотой менее 1:100 000 – редкие заболевания.
Дата добавления: 2018-11-24; просмотров: 979; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!