Основные подходы к терапии и ведение больных с первичными иммунодефицитными состояниями.
Основными задачами ведения больных с ПИДС являются:
1) Коррекция имеющегося иммунологического дефекта
2) Профилактика и/или адекватная терапия инфекционных проявлений
3) Раннее выявление и терапия неинфекционных проявлений
4) Социальная адаптация детей с ПИДС.
Несмотря на то, что больные с первичными иммунодефицитом проходят специальное обследование и терапию в специализированных стационарах, основная роль в выявлении этих состояний принадлежит врачам первичного звена. В связи с этим Европейское и Панамериканское общества иммунодефицитов (ESID PAGID) предложили использовать следующие критерии риска ПИДС (Табл.2). При наличии одного или нескольких из этих признаков больному показано проведение иммунологического обследования и консультация специалиста-иммунолога.
12 НАСТОРАЖИВАЮЩИХ ПРИЗНАКОВ ПЕРВИЧНОГО ИММУНОДЕФИЦИТА У ДЕТЕЙ И ВЗРОСЛЫХ
Другие статьи
. Чем настоящий иммунодефицит отличается от частой простуды
Почему дети должны болеть, о бессмысленности иммуномодуляторов и о том, чем настоящий иммунодефицит отличается от частой простуды, — иммунолог Анна Щербина Иммунодефицит — это состояние, сопровождающееся значительными и долговременными изменениями в иммунной системе и серьезными симптомами. Есть иммунодефициты вторичные, а есть первичные (ПИД). Первичные обусловлены генетически. Как правило, симптомы возникают в раннем возрасте, однако иногда могут возникнуть и у взрослых. Но в любом случае проявления будут очень тяжелыми. С первичными иммунодефицитами встречаются крайне редко. Подтвердить многие такие заболевания можно, обнаружив дефект гена. Но пока, правда, найдены мутации не при всех ПИД, поиск продолжается. Текст: Дарья Саркисян Фотографии: Максим Шер Журнал "Большой город"
1 февраля 2018
|
|
|
ЧТО ТАКОЕ ПЕРВИЧНЫЙ ИММУНОДЕФИЦИТ
Что такое Первичный иммунодефицит, как передаётся, как часто встречается, какие есть формы?
1 февраля 2018
Как рассказать детям об иммунитете
Презентация для школьников о том, что такое иммунитет, какие нарушения встречаются, как живут дети с первичным иммунодефицитом и как им можно помочь.
24 ноября 2015
Первичный иммунодефицит. Х-сцепленный лимфопролиферативный синдром
Х-сцепленный лимфопролиферативный синдром – первичный иммунодефицит, при котором у пациентов мужского пола отмечается нарушение иммунного ответа на вирус Эпштейн-Барра.
6 июня 2014
Первичный иммунодефицит. Аутоиммунный лимфопролиферативный синдром
Аутоиммунный лимфопролиферативный синдром – первичный иммунодефицит, при котором отмечается хроническое незлокачественное увеличение лимфоузлов, печени и селезенки, аутоиммунная патология, повышение уровня иммуноглобулинов в крови.
6 июня 2014
|
|
|
Первичный иммунодефицит. Синдром Ди Джорджи
Синдром Ди Джорджи – врожденный дефект, который приводит к гипоплазии или отсутствию тимуса (вилочковой железы) в сочетании с пороками развития крупных сосудов, сердца, паращитовидных желез, костей лицевого черепа и верхних конечностей
6 июня 2014
Оптимизация диагностики и терапии наследственного ангионевротического отека у взрослых.
Особенности редкой формы первичного иммунодефицита, клинические проявления, иммунологические нарушения и принципы терапии наследственного ангионевротического отека. Индивидуальные планы самоконтроля для каждого пациента и оценкаа их эффективность. Караулов А.В., Сидоренко И.В., Капустина А.С. Первый Московский государственный медицинский университет им. И. М. Сеченова, Москва
20 мая 2014
Наследственный ангионевротический отек
Наследственный ангионевротичекий отёк - редкое, жизнеугрожающее заболевание, которое относится к группе первичных иммунодефицитов. Причина - недостаточность общего уровня или снижение функциональной активности С1-ингибитора системы комплемента. Жизнь таких больных становится кошмаром: они никогда не знают, где и когда начнется отек. Пациенты нередко испытывают страх очередного приступа, для них характерны чувство одиночества, ощущение безысходности и бесконечные проблемы на работе, в учебе и быту.
20 мая 2014
|
|
|
Первичный иммунодефицит. Хроническая гранулематозная болезнь
Хроническая гранулематозная болезнь (ХГБ) – генетическое заболевание, связанное с дефектом фагоцитов, клеток иммунной системы, которые защищают организм путем поглощения (фагоцитоза) вредных чужеродных частиц, бактерий, а также мертвых или погибающих клеток, из-за которого снижается их антимикробная активность.
13 марта 2013
Первичный иммунодефицит. ОВИН - общая вариабельная иммунная недостаточность
Общая вариабельная иммунная недостаточность - нарушение, характеризующееся низкими уровнями иммуноглобулинов (антител) в сыворотке крови и повышенной чувствительностью к инфекциям. Данная статья предназначена для пациентов и членов их семей и не должна заменять совета клинициста-иммунолога.
27 июля 2011
Первичный иммунодефицит. Синдром Вискотта-Олдрича
Синдром Вискотта-Олдрича является первичным иммунодефицитным состоянием, поражающим как Т- лимфоциты, так и В-лимфоциты. Также тяжело поражаются тромбоциты - клетки, помогающие останавливать кровотечение. Информация в статье предназначена для пациентов и членов их семей и не должна заменять рекомендаций и назначений лечащего врача и клинициста-иммунолога.
27 июля 2011
|
|
|
Первичный иммунодефицит. Х-сцепленная агаммаглобулинемия
У больных с Х-сцепленной агаммаглобулинемией основным дефектом является неспособность предшественников В-лимфоцитов созревать до состояния В-лимфоцитов, а затем плазматических клеток. Поскольку у этих больных нет клеток, вырабатывающих иммуноглобулины, наступает тяжелая недостаточность иммуноглобулинов. Информация в статье предназначена для пациентов и членов их семей и не должна заменять рекомендаций и назначений лечащего врача и клинициста-иммунолога.
18 июля 2011
Первичный иммунодефицит. ТКИН - тяжелая комбинированная иммунная недостаточность
Тяжелая комбинированная иммунная недостаточность (ТКИН) - самый тяжелый диагноз в списке первичных иммунодефицитов - является редким синдромом, обусловленным различными генетическими факторами, и сочетающим отсутствие функций Т- и В- лимфоцитов (а во многих случаях также отсутствие функции естественных киллеров или NK-лимфоцитов). Эти нарушения приводят к чрезвычайной чувствительности к тяжелым инфекциям. Информация в статье предназначена для пациентов и членов их семей и не должна заменять рекомендаций и назначений лечащего врача и клинициста-иммунолога.
18 июля 2011
12 настораживающих признаков первичного иммунодефицита
ПИД не СПИД. Первичный иммунодефицит является врожденным нарушением в иммунной системе, имеющим генетическую природу. Показанием для направления к иммунологу является сочетание рецидивирующих вирусных и бактериальных инфекций либо наличие тяжелых, затяжных бактериальных инфекций. Данные Всемирной организации здравоохранения свидетельствуют о том, что частота ОРВИ 8 раз в год является нормальным показателем для детей дошкольного и младшего школьного возраста, посещающих детские учреждения.
29 июня 2011
Часто болеющие дети: чем они больны на самом деле?
Инфекции уха, горла, носа, а также бронхолёгочные инфекции составляют основной перечень заболеваний в детском возрасте. Данные ВОЗ свидетельствуют о том, что частота ОРВИ 8 раз в год является нормальным показателем для детей дошкольного и младшего школьного возраста, посещающих детские учреждения. Показанием для направления к иммунологу является сочетание рецидивирующих вирусных и бактериальных инфекций либо наличие тяжелых, затяжных бактериальных инфекций.
29 июня 2011
Иммунодефициты у детей.
Диагноз «иммунодефицит» становится все более популярным у врачей разных специальностей. Создается впечатление, что зачастую врачи, вместо того, чтобы четко определить диагноз и проводить лечение заболевания в соответствии с утвержденными стандартами, назначают иммунотропные средства, не представляя эффект и последствия такой терапии.
29 июня 2011
Диагностика семей с иммунодефицитом
Первичные иммунодефициты, являются наследуемыми заболеваниями, при которых родители являются носителями больного гена и передают его детям. В результате чего у ребенка развивается заболевание. В настоящее время в связи с развитием генетики и иммунологии известны многие гены, мутация в которых приводит к развитию различных форм первичных иммунодефицитов.
10) Принципы диагностики и иммунотерапии больных первичными иммунодефицитами.
ДА ВЫ РОФЛИТЕ КНИЖКУ СКРИНИТЬ!!!!!





11) Тяжелые комбинированные иммунодефициты (ТКИД), основные варианты. Клиника, диагностика, подходы к лечению.
Тяжёлый комбинированный иммунодефицит— это генетическое заболевание, при котором в результате дефекта одного из генов нарушается работа компонентов адаптивной иммунной системы B- и T-лимфоцитов.
Наиболее часто цитируемый показатель распространённости тяжёлого комбинированного иммунодефицита составляет примерно 1 на 100,000 родившихся.
| Тип | Описание |
| X-сцепленный тяжёлый иммунодефицит | Наиболее распространённый тип тяжёлого комбинированного иммунодефицита, возникающий из-за мутаций в гене, кодирующем общие гамма-цепи, белок которых является общим для рецепторов интерлейкинов IL-2, IL-4, IL-7, IL-9, IL-15 и IL-21. Перечисленные интерлейкины и их рецепторы вовлечены в процессы развития T- и B-лимфоцитов. В результате мутаций происходят дисфункции общей гамма-цепи, и, как следствие, дефект распространяется на процесс сигнализации интерлейкина. Происходит почти полный отказ иммунной системы как со стороны развития, так и со стороны функционирования, с отсутствием или очень малым количеством T-лимфоцитов, NK-клеток и нефункциональными B-лимфоцитами. Общая гамма-цепь кодируется геном IL-2 рецепторов гамма, который находится на X-хромосоме. Наследуется как рецессивный признак. |
| Дефицит аденозиндеаминазы | Второй по распространённости тип тяжёлого комбинированного иммунодефицита. Его причиной является дефект фермента аденозиндеамиазы, который необходим для расщепления пуринов. Недостаток аденозиндеаминазы провоцирует накопление dATP. Этот метаболит ингибирует активность фермента рибонуклеотидредуктазы, участвующего в превращении рибонуклеотидов в дезоксирибонуклеотиды. Эффективность иммунной системы зависит от пролиферации лимфоцитов и, следовательно, синтеза dNTP. Если рибонуклеотидредуктаза не способна нормально функционировать, пролиферация лимфоцитов блокируется, а иммунная система компрометируется. |
| Синдром Оменна | Производство иммуноглобулинов требует участия рекомбинантного фермента, полученногог от рекомбинации генов, активирующих RAG-1 и RAG-2. Эти ферменты участвуют в первом этапе V(D)J рекомбинации, в котором сегменты B-лимфоцитов или ДНК T-лимфоцитов перестраиваются, создавая новые T- или B-клеточный рецепторы. Некоторые мутации RAG-1 или RAG-2 продотвращают процесс V(D)J рекомбинации, тем самым приводя к возникновения ТКТД. |
| Синдром голых лимфоцитов | MHC класса II не экспрессируется на поверхности антигенпредставляющих клеток. Аутосомно-рецессивный тип наследования. |
| Дефицит JAK3 | JAK3 является ферментом, который выступает посредником трансдукции через общую гамма-цепь. Мутация гена JAK3 также вызывает тяжёлый комбинированный иммунодефицит. |
| Дефицит DCLRE1C/Artemis | Несмотря на то, что исследователями было идентифицировано около дюжины генов, вызывающих ТКИД, население Навахо и Апачи страдает наиболее тяжёлой формой заболевания. Это связано с отсутствием гена DCLRE1C/Artemis. Без этого гена организм ребёнка не в состоянии восстановить ДНК или вырабатывать антитела. |
(Симптомы) Иммунодефициты характеризуются различным набором клинических признаков. Проявлениями этого заболевания всегда являются тяжелые инфекции. К таким инфекциям относятся: пневмония; менингит; инфекции крови. Другие симптомы: воспалительные процессы слизистой; увеличение лимфоузлов; кожные поражения (сыпь, язвы); нарушения функции печени, почек; респираторные симптомы; молочница (грибковые инфекции полости рта и половых органов); диарея; рвота; ферментные нарушения; аллергические проявления; кашель, хрипы; плохие показатели анализов крови.
(Диагностика) Некоторые формы тяжёлого комбинированного иммунодефицита могут быть обнаружены путём секвенирования ДНК плода, если есть основания подозревать данное заболевание. В противном случае, наследственное заболевание не диагностируется примерно до 6 месяцев. Как правило, на его наличие могут указывать рецидивирующие инфекции. Задержка в обнаружении тяжёлого комбинированного иммунодефицита обусловлена тем, что у новорожденных в течение первых нескольких недель жизни присутствуют антитела матери, и дети с с таким иммунодефицитом выглядят здоровыми.
(Лечение) Наиболее распространённым методом лечения тяжёлого комбинированного иммунодефицита является трансплантация гемопоэтических стволовых клеток, которая проходит успешно либо при участии неродственного донора, либо при участии полу-совместимого донора, которым может являться один из родителей.
(Лечение) Также врачи успешно проводили внутриутробную трансплантацию, сделанную до рождения ребёнка, с использованием пуповинной крови, богатой стволовыми клетками.
12) Иммунодефициты с нарушением антителообразования. Клиника, диагностика, подходы к лечению.



13) Первичные иммунодефициты с дефектом фагоцитарного звена.
Хроническая гранулематозная болезнь
Наследственное заболевание, обусловленное дефектом генов NADPH-окси-дазы. Описаны мутации генов, кодирующих 4 субъединицы NADPH- оксидазы — gp91, p22, p47, p67. Мутации в этот ген ведут к развитию Х-сцепленной хронической гранулематозной болезни. Остальные варианты хронической гранулематозной болезни — аутосомно-рецессивные.
Основное проявление заболевания — нарушение (полное или частич- ное) образования активных форм кислорода и, вследствие этого, ослаб- ление бактерицидной активности фагоцитов, приводящее к сохранению жизнеспособности фагоцитированных патогенов. Клинически синдром проявляется на первом году жизни тяжелыми рецидивирующими бактери- альными и грибковыми инфекционными заболеваниями. В первую очередь поражаются органы, контактирующие с внешней средой — легкие, органы пищеварения, кожа, а также дренирующие их лимфоузлы. Позже вследствие гематогенного распространения патогенов поражаются печень, мозг, кости, почки. Часто возбудителями при таких заболеваниях служат грамположи- тельные бактерии: Streptococcus aureus, Aspergillus, Escherichia сoli.
В основе иммунодиагностики хронической гранулематозной болезни лежит идентификация продуктов кислородного взрыва. Для этого исполь- зуют NBT-тест (оценка восстановления красителя нитросинего тетразолия), хемилюминесцентное определение активных форм кислорода и цитометри- ческое определении внутриклеточной перекиси водорода (по индукции обра- зования флуоресцирующего метаболита дихлорфлюоресцеиндиацетата).
Синдром Чедиака–Хигаси (Chediak–Higashi)
Редкое аутосомно-рецессивное заболевание, характеризующееся тяже- лым иммунологическим дефектом, рецидивирующими бактериальными инфекциями, а также частичным альбинизмом глаз и кожи. Характерный диагностический признак — наличие гигантских гранул в нейтрофилах, CD8+ Т- и NK-клетках, меланоцитах и тромбоцитах. Гигантские гранулы являются видоизмененными лизосомами, цитолитическими гранулами, меланосомами и плотными тельцами тромбоцитов.
Идентифицирован ген CHS/Beige, мутации которого ответственны за раз- витие данного синдрома, а мутации его гомологов — за развитие патологии у мышей beige и алеутских норок. Ген кодирует белок, обеспечивающий транспорт секреторных везикул. Мутации, приводящие к образованию гигантских гранул, вызывают формирование стоп-кодона и синтез уко- роченной формы белка. Основной эффект мутации состоит в нарушении внутриклеточного транспорта и экзоцитоза гранул. Отсутствие выделения меланина из гигантских гранул считают причиной альбинизма.
В иммунной системе сильнее всего страдают связанные с лизосомальны- ми гранулами функции клеток — нейтрофилов (нарушается их фагоцитар- ная активность), NK-клеток и цитотоксических Т-лимфоцитов (ослаблена их способность к перфоринзависимому цитолизу). Таким образом, главный иммунологический дефект при синдроме Чедиака–Хигаси состоит в нару- шении функций, связанных с транспортом лизосомальных гранул в клетках иммунной системы.
Дефекты миелопероксидазы и глюкозо-6-фосфат дегидрогеназы
Следствие этих нарушений — снижение образования активных форм кислорода и внутриклеточного киллинга бактерий из-за нарушения функ- ций названных ферментов.
Описано еще несколько очень редких синдромов, затрагивающих фаго- циты и их функции: синдром Грисчелли (дефект секреторных лизосом, их дегрануляции и опосредуемого ими цитолитического действия), синдром Германски–Пудлак (дефект секреторных гранул и тромбоцитов), синд- ром Швахмана–Диаманд (нарушение хемотаксиса нейтрофилов), синдром Барта (нейтропения).
14) Вторичные иммунодефициты, характеристика, причины развития, патогенетические механизмы развития, иммунодиагностика, клинические проявления, подходы к лечению.
СЛАВА - ТЫ АХУЕНЕН , В СЛЕД РАЗ, ПРОСТО ССЫЛКУ НА УЧЕБНИК СО СТРАНИЧКАМИ КИДАЙ ))))))00))






15) Синдром приобретённого иммунодефицита (СПИД). Определение. Этиология. Природа вируса иммунодефицита человека (ВИЧ), пути трансмиссии.
|
| |
|
| |
Приобретенная форма ВИД –
синдром приобретенного иммунодефицита, развивающийся на фоне ВИЧ-инфекции
ВИЧ-инфекция – инфекционное заболевание, вызванное вирусом иммунодефицита человека (human immunodeficiency virus infection – HIV-infection).
Синдром приобретенного иммунодефицита (СПИД) – клиническое состояние, развивающееся на фоне ВИЧ-инфекции, характеризующееся падением числа CD4+ Т-лимфоцитов, развитием оппортунистических инфекций и опухолей, и приводящее к гибели инфицированного человека
По классификации не относится ни к индуцированным, и к спонтанным формам вторичных иммунодефицитов. Есть 2 вида вирусов, отличающихся по содержанию генома: ВИЧ-1 - имеет ген vpu, а ВИЧ-2 содержит vpx. Второй распространен в Западной Африке и менее патогенен. Первый в США, России, Европе и Центральной Африке.
Этиология: причиной данного заболевания является наличие в организме вируса иммунодефицита человека (ВИЧ).
Природа возбудителя: вирус иммунодефицита человека принадлежит к семейству РНК — содержащих ретровирусов (особенность — наличие фермента обратной транскриптазы), к подсемейству так называемых медленных лентивирусов.
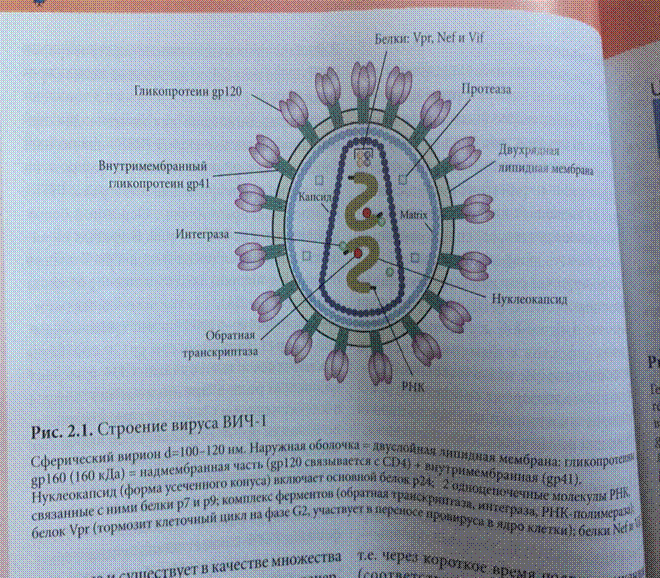
Основной белок нуклеокапсида ВИЧ — р24. В нуклеотиде заключены две одноцепочечные молекулы РНК, связанные с ними белки р7 и р9, комплекс ферментов (обратная транскриптаза, интеграза, РНК-полимераза, протеиназа). В состав оболочки вируса входят гликопротеины gp160, состоящие из надмембранной части gp120 и внутримембранной — gp41. Гликопротеин gp120 способен связываться с молекулами СD4 и играет ключевую роль в проникновении вируса в клетку-мишень.
Пути трансмиссии:
Источник — ВИЧ-инфицированные люди в любой период течения инфекционного процесса. Вирус обнаруживают во всех биологических жидкостях заражѐнного человека, но в разных титрах: максимальная концентрация в крови, сперме, вагинальном отделяемом, грудном молоке.
Пути заражения:
- парентеральный (переливание инфицированной крови и еѐ компонентов, плазмы, трансплантация органов, искусственное оплодотворение, инъекционное введение наркотиков, через инфицированные медицинские инструменты)
- половой (риск тем выше,чем больше половых партнеров)
- вертикальный (от матери новорожденному): антенатальный вариант (через плаценту, вероятность инфицирования 15 — 25%), интранатальный (в родах, 60-85%) , постнатальный (при грудном вскармливании, 12 — 25% случаев). При повторных беременностях риск возрастает в 2-3 раза.
16) Нарушения в иммунной системе при СПИДе.
Иммунодефицит, вызываемый ВИЧ-1, обусловлен в первую очередь уменьшением количества и ослаблением функций СD4 Т-лимфоцитов. Происходит аномальная активация В-лимфоцитов. Развивается гипергаммаглобилинемия и увеличивается образование IgG и IgA, в большинстве случае специфичных к ВИЧ-1. Нарушается синтез цитокинов. Уменьшается содержание ИФНгамма, снижается уровень ИЛ2, секретируемого Т-лимфоцитами и регулирующего пролиферацию и дифференцировку Т- и Влимфоцитов, а также уменьшается содержание других цитокинов (ИЛ-12, ИЛ-13, ИЛ-16). Мононуклеарные фагоциты образуют повышенное количество ФНОа, что способствует прогрессированию заболевания.
17) Лабораторные методы диагностики ВИЧ-инфекции.
Серологический метод является наиболее распространенным и доступным. Он заключается в поиске АТ к АГ ВИЧ.
Антитела появляются через 1-3 месяца после инфицирования и обнаруживаются на всех стадиях. При развитии СПИДА их титр снижается.
Основной диагностической реакцией является ИФА. Разработаны специальные тест-системы и постановка реакции автоматизирована.
В начале обычно проводят скрининговое исследование, затем в случае положительного результата, который считается предварительным, проводят подтверждающее исследование. Его проводят методом иммуноблотинга, выявляя наличие антител к gp120 и gp41, а также белкам p24 p31.
Вирусологическое исследование является трудоемким и дорогостоящим методом и клинической практике практически не используется.
Молекулярно-генетический метод. Используют ПЦР для определения провирусной ДНКв крови. ПЦР дает возможность обнаружить ВИЧ уже через 3 недели после заражения.
18) Основные принципы иммунотерапии.




19) Метод выделения мононуклеарных клеток из периферической крови человека.
Мононуклеарные клетки выделяют (рис. 2.2, см. также цв. вклейку) из периферической крови человека по методу Boyum (1968), основанному на седиментации в одноступенчатом градиенте плотности фиколл-урографина. Фиколл в данной смеси выступает как агент, агрегирующий эритроциты, а изопак (или урографин) нужен для создания изотоничности и плотности 1,077 г/см3.
Принцип метода.Гепаринизированную кровь разводят в 3 раза культуральной средой и аккуратно наслаивают на градиент фиколлурографина. Кровь задерживается над фиколлом и не смешивается с ним. Постепенно эритроциты склеиваются фиколлом и опускаются на дно пробирки. Гранулоциты, имеющие плотность большую, чем седиментирующий раствор, оседают вместе с эритроцитами. Лимфоциты вместе с моноцитами остаются в интерфазе, их собирают, переносят в другую пробирку и отмывают центрифугированием.
Метод не дает выхода более 90% клеток.
 Рис. 2.2.Выделение мононуклеарных клеток в одноступенчатом градиенте плотности фиколл-урографина
Рис. 2.2.Выделение мононуклеарных клеток в одноступенчатом градиенте плотности фиколл-урографина
20) Проточная цитофлюориметрия. Принцип метода, возможности использования.
Основная идея проточной цитометрии — «поштучный» анализ клеток в потоке, проходящий с большой скоростью. Представьте себе суспензию клеток, которую необходимо изучить: это может быть кровь или ее фракции, отдельные популяции лимфоцитов или даже потенциально раковые клетки. Если это клеточная линия, то задача достаточно тривиальная — вы можете считать, что суспензия однородная, то есть все клетки одинаковые просто по определению клеточной культуры . Но что, если вам нужно изучить неоднородную суспензию — например, в вашей клеточной линии наблюдается самопроизвольная дифференцировка? Или же вы хотите исследовать биологические жидкости (например, кровь), где находится несколько (или даже много) типов клеток? А что, если вам нужно отличить раковые клетки от здоровых?
Особенно важно, что иногда отличить разные клетки при помощи микроскопии не представляется возможным: в микроскоп они просто-напросто выглядят одинаково. А ведь нам зачастую нужно не просто отличить одни клетки от других, но и проанализировать те или иные их параметры: выживаемость, экспрессию белков, целостность ДНК и многое другое.
В этом случае на помощь и придет проточная цитометрия. Ее физические принципы весьма просты. Суспензия клеток, предварительно помеченных светящимися молекулами (флуорохромами), помещается в поток жидкости, пропускаемый через проточную ячейку. Получается как бы «поток в потоке», что порождает эффект гидродинамического фокусирования: исследуемые клетки выстраиваются в цепочку и в таком порядке пересекают пучок световых (обычно лазерных) лучей, служащих для анализа каждой отдельно взятой клетки.
Свет, исходящий от флуорохромов, фокусируют при помощи оптической системы, состоящей из нескольких зеркал и линз, а затем раскладывают на определенные компоненты. Полученные световые сигналы преобразуют в электрические импульсы и анализируют при помощи специального программного обеспечения.
Таким образом, результат цитометрического анализа — определение состояния каждой клетки в каждой из популяций образца. Всего за несколько десятков секунд сквозь проточную ячейку проходят тысячи клеток, позволяя исследователю сделать вывод о составе и характеристиках клеточной суспензии. Также при помощи проточного цитометра можно определить абсолютное число клеток в исследуемом образце с использованием калибровочных микросистем.
Основные принципы метода
Суть проточной цитометрии, пожалуй, можно изложить всего одним предложением: поток клеточной суспензии в ячейке сжимается, клетки выстраиваются в очередь и проходят через лазерный луч, регистрация рассеяния от которого позволяет охарактеризовать каждую клетку в потоке индивидуально.
Клетки, исследуемые цитометрией, метят флуорохромами, поэтому лазерный луч возбуждает вторичное свечение (флуоресценцию). Затем сигналы флуоресценции и светорассеяния регистрируются различными детекторами. Информация от детекторов используется компьютерным алгоритмом, который позволяет в наглядной форме «посчитать» клетки и разделить их на отдельные популяции, отличающиеся друг от друга по каким-то важным параметрам, интересующим исследователя.
В зависимости от используемых флуорохромов требуются разные длины волн для их возбуждения и разные детекторы (фильтры) для анализа испускаемого свечения. В современных приборах может быть до 7 лазеров и до 30 каналов детекции, но для большинства приложений достаточно 1–3 лазеров и 4–10 каналов.
Проточная цитометрия в иммунологии активно используется для иммунофенотипирования клеток крови, позволяет идентифицировать внутриклеточные белки, оценить степень цитотоксичности.
Дата добавления: 2019-07-15; просмотров: 276; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!