Нарушение количественного поступления белка.
Избыточное поступление белков возможно при переедании, несбалансированной диете, сахарном диабете.
Избыточное потребление белка вызывает положительный азотистый баланс. Часть избыточного белка расходуется в глюконеогенезе, увеличивая теплопродукцию, часть задерживается в виде циркулирующих аминокислот. Перекорм белками не вызывает ожирения. При значительном избытке пищевого белка создается повышенная нагрузка на печень и почки, так как необходимо нейтрализовать дополнительного аммиака и выведения мочевины. Возникают диспепсические расстройства, дисбактериоз с развитием кишечной аутоинфекции и отвращение к белковой пище.
Белково-калорическая недостаточность возможна при голодании.
крайние состояния: квашиоркор и алиментарный маразм.
Квашиоркор – несбалансированная алиментраная недостаточность белка. (снижение массы тела, гипопротеинемия, гиполипопротеинемия, отрицательный азотистый баланс, тотальные отеки, асцит, иммунодефициты, гиперальдостеронизм (результат гиповолемии), задержка физического и умственного развития) прогноз неблагоприятный из-за атрофии тонкой кишки и ахилии, смертность высокая.
Алиментарный маразм – сбалансированная белково- калорическая недостаточность. Полное или частичное белковое голодания приводит к мобилизации белка из костей, мышц, кожи (снижение массы тела, отрицательный азотистый баланс, гипогликемия, кетонемия, кетоацидоз, гиперкортицизм, повышение уровня глюкагона и соматостатина, гиперкалиемия, иммунодефицит, задержка развития) Прогноз благоприятный.
|
|
|
Нарушение аминокислотного состава потребляемого белка.
Белки пищи содержат 22 АК, 8 из них незаменимы (метионин, лизин, триптофан, фенилаланин, лейцин, изолейцин, треонин, валин). У новорожденных 10 незаменимых АК (гистидин, аргинин)
Дефицит любой из незаменимых АК ведет к:
- отрицательному азотистому балансу (усилен катаболизм эндогенных белков)
-замедлению роста и развития
-снижению регенераторной активности тканей и органов
- уменьшение массы тела
- снижение аппетита и усвоения белка пищи
Нарушение переваривания белков.
Нарушение расщепления белка в желудке и переваривания его в тонкой кишке.
Нарушение расщепления белка в желудке при гипоацидных состояниях, снижение содержания или активности пепсина, резекция желудка.
Последствия: нарушение набухания белка, торможение переваривания коллагенового компонента, недостаточное расщепление белков мышечных волокон, замедление эвакуации пищи в 12-персную кишку.
Нарушения переваривания белка в тонкой кишке при синдроме мальабсорбции и нарушения переваривания белка в кишечнике.
|
|
|
Креаторея- присутствие в кале непереваренных или полупереваренных мышечных волокон при первичной или вторичной панкреатической недостаточности, при синдроме Цоллингера-Эллисона (инактивация кишечного содержимого из-за быстрой эвакуации желудочного секрета)
При очень выраженной недостаточности желудочного и панкреатического пищеварения креаторея сменяется лиентореей (крупные комки непереваренной пищи в фекалиях)
Целиакия – синдром, характеризующийся нарушением полостного и пристеночного пищеварения, торможением всасывания АК
Недостаточность энтерокиназы – нарушается активация трипсина и других пептидаз-гидролаз полостного пищеварения. Резко падает протеолитическая активность кишечного содержимого. Развивается эндогенное белковое голодание организма, гипотрофия, гипопротеинемия, безбелковые отеки.
Кишечная аутоинтоксикация- нарушение самочувствия и функций внутренних органов с запорами и гнилостной диспепсией. При нарушении переваривании белков и всасывания АК продукция токсических метаболитов микрофлорой кишечника резко возрастает. Это кадаверин, гистамин, октопамин, тирамин, пирролидин, пиперидин, диметиламин, серотонин, путресцин, из триптофана, под влиянием триптофаназы Е coli. Образуются индол, скатол, скатоксил, индоксил. Из тирамина и тирозина- фенол крезол. Эти токсины через портальную вену поступают в печень, где обезвреживаются и затем экскретируются с мочой. Но при увеличенном объеме токсических метаболитов и исчерпания дезинтоксикационной ф. печени приводят к патологическим последствиям. При кишечной аутоинтоксикации продуктами гниения белков наступают колебания АД, понижение болевой чувствительности, анемия, миокардиодистрофия, понижение аппетита, нарушение желудочной секреции, в тяжелых случаях угнетение дыхания, сердечной деятельности, кома.
|
|
|
Азотистый баланс – разность между количеством азота, поступившего в организм и количеством азота, выделенного организмом во внешнюю среду. У здорового взрослого человека азотистый баланс нулевой.
| Положительный азотистый баланс (количество поступающего азота больше количества выделяемого) | Отрицательный азотистый баланс (количество выделяемого азота больше количества поступающего) |
| Рост Регенерация Лактация и беременность | старение |
| Полицитемия Крупные доброкачественные опухоли Гиперсекреция гормона роста | Голодание Белково-энергетическая недостаточность ИЗСД Гиперкортицизм стресс |
|
|
|
94. Наследственные нарушения аминокислотного транспорта. Этиология, патогенез, последствия. Нарушения чрезмембранного транспорта аминокислот. Гастроэнтерологические и нефрологические аспекты. Аминоацидурия.
Трансмембранный перенос аминокислот определяет 3 пункта: всасывание их в кишечнике, захват их печенью, реабсорбцию почками (так же и в других системах – желчном пузыре, мозге, скелете, мышцах, эритроцитах). Механизм при этом один: энергозависимый транспорт с участием иона Na по градиенту концентрации без затраты АТФ (но есть и другие механизмы транспорта аминокислот в клетку, но все они вторичны). При адекватном переваривании всасывание аминокислот порядка 98 процентов.
При всасывании кишечником участвуют пермеазные системы, таких систем пять – они работают с определенными группами химически близких аминокислот: 1) транспорт относительно крупных нейтральных моноаминокарбоновых аминокислот 2) Транспорт двуосновных аминокислот (орнитин, аргинин, лизин) и цистеина 3) перемещение кислых дикарбоновых аминокислот (аспарагиновой, глутаминовой). 4) наиболее малых по размеру: глицина, оксипролина, пролина. Так же существует мнение о наличии индивидуальных переносчиков. Так же есть система внутриклеточного переноса коротких пептидов (последующий внутриклеточный гидролиз, дополняющий аминокислотный транспорт). Так же аминокислоты конкурируют за одну и ту же транспортную систему.
Аминоацидопатии – нарушения транспорта и интермедиарного обмена аминокислот. Всего таких описано 10 (из них 5 – вызваны аномалиями группоспецифических транспортеров (пермеаз) и вовлекают каждая несколько близких по строению аминокислот.
Цистинурия, болезнь Хартнупа и двухосновная аминоацидурия – клинически значимые проявления, а дикабоксиламиноацидурия и иминоглицинурия – бессимптомны!!!
Другие 5 (гиперцистинурия, гистилинурия, лизинурия, мальабсорбция триптофана и метионина) – субстрат – специфичные.
Все описанные состояния – аутосомно-рецессивные многогенные наследственные болезни.
Цистинурия (самая встречаемая и клинически важная). Цистин – малорастворим. Он формирует камни в почках, мочевом пузыре, уретре. Имеет три типа: 1 – нет выраженной гипераминоацидурии, 2 – мочевые симптомы + снижено кишечное всасывание, увеличена почечная экскреция орнитина, лизина, аргинина, 3 – нарушение почечной, интактной кишечной абсорбцией аминокислот. Нагрузка пищевая цистеином не ведет к ухудшению мочевых симптомов, так как эта аминокислота плохо всасывается в кишечнике. + наследственная гиперцистеинурия (изолированно нарушен транспорт цистеина).
Болезнь Хартнупа – нарушено всасывание и увеличена экскреция крупномолекулярных нейтральных аминокислот, вторично (не хватает триптофана) раз-ся нарушения синтеза НАД.
Дибазикаминоацидурии – не реабсорбируется аргинин, орнитин, лизин (но не цистеин – не хар-но камнеобразование). Нарушается цикл мочевины, гипераммониемия, задержка роста. Умственное отставание.
Мальабсорбция триптофана – ведет к усиленному образованию в кишечнике производных индола. Поражение нервной системы (нарушается кроветворение). Кальциноз почек.
Метиониновая мальабсорбция – умственное отставание, задержка роста, гипопротеинемические отеки, нарушенное образование меланина.
Лизинурия – судороги, нарушение психомоторного развития.
Гистидинурия – нарушения синтеза гемоглобина и функций ЦНС.
Оксопролинурия – не расщепляется пирролидонкарбоновая кислота (побочный продукт при отщеплении аминокилот, переносимых в клетки, от глутатионового транспортера.
Синдром Лоува – наследственное, сцепленное с Х-хромосомой. Генерализованный дефект транспорта аминокислот. +ацидоз, задержка физического и психомоторного развития, двусторонняя глаукома и катаракта. Гипотонус мышц, черепная дизморфия.
Синдром Фанкони – аминоазот возрастает в мочи в 30-40 раз. Канальцевый ацидоз, полиурия с выведением всех аминокислот.
Синдром Людера-Шелдона – аминоцидурия без поражения без поражения кишечноц абсорбции аминокислот, сопровождается фосфатурией.
Синдром Роули-Розенберга – задержка физического развития, гипоплазия мышц, поражение легких, правожелудочковая сердечная недостаточность, дефект почечной реарбсорбции всех аминокислот.
Прибретенные нарушения транспорта: отправления тяжелыми металлами. Эндогенное отравление происходит при болезни Коновалова-Вильсона.
При наследственных нарушениях активации и рецепции витамина D, так же нарушается реабсорбция ряда аминокислот, из-за ингибирующего действия парат-гормона на этот процесс при вторичном гиперпаратиреозе.
До 3-х месяцев жизни система переноса еще не развивается в полную силу, есть место физиологической преходящей аминоацидурии новорожденных.
Аминоацидурия – 99% аминокислот должны реабсорбироваться тубулярным аппаратом почек. Следовательно, аминоацидурией или гипераминоацидурией называется такое состояние, когда с мочой выводится избыточное количество аминокислот и промежуточных продуктов их обмена.
Есть три группы основных механизмов возникновения данного состояния:
- повышается концентрация аминокислот в крови выше максимальных возможностей почечной реабсорбции (допустим, перекорм белками). В этой же группе – наследственные илиприобретенные нарушения процессов дезаминирования и переаминирования в печени.
- конкурентное ингибирование одной аминокислотой других аминокислот (такая аминоацидурия называется смешанной).
-дефект транспортера или сопряженного с ним энергетического процесса в самих канальцах.
Часто аминоацидурия сопровождается гипераминоацидемией.
95. Наследственные нарушения межуточного обмена аминокислот. 1Фенилкетонурии, 2алкаптонурия, 3лейциноз, 4гомоцистинурия, нарушение 5обмена тирозина. Этиология, патогенез, механизмы основных проявлений.
Наследственные нарушения межуточного обмена АК (аминоацидопатии) вызваны различными мутациями. В норме наиболее велика скорость обмена белков в нервной ткани. Поэтому наследственные аминоацидопатии - одна из причин слабоумия.
1 Фенилкетонурия (или фенилпировиноградная олигофрения) – мозаичная аминоацидопатия, мутация в 12 хромосоме, аутосомно-рециссивная, имеет несколько генокопий. В большинстве случаев сопровождается дефицитом печеночного фермента фенилаланин-4-гидроксилазы, что ведет к резкому увеличению фенилаланина в крови.
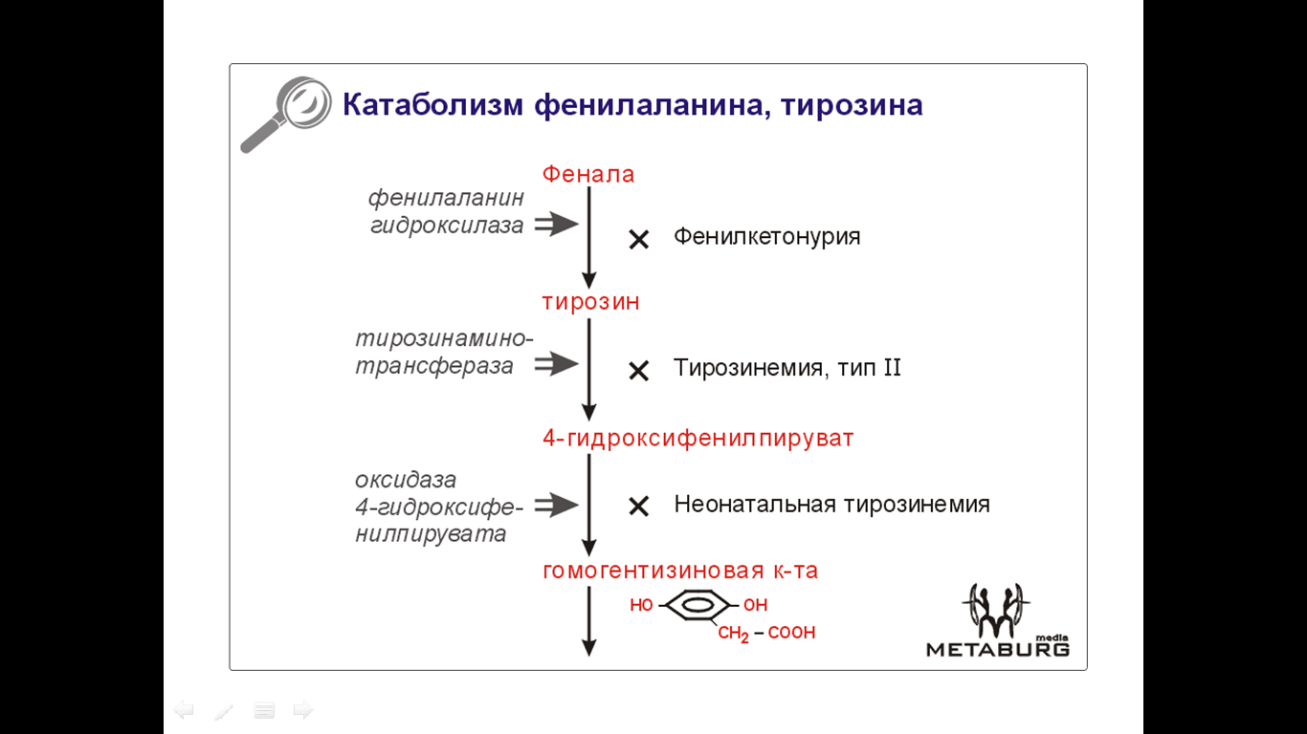
Недостаток превращения фенилаланина в тирозин, подавление избытком фенилаланина активности тирозиназы ведут к дефициту тирозиновых и триптофановых производных (меланина=>светлые кожа и глаза; катехоламинов=>гипотензия; серотонина=>судороги и тремор).

Избыток фенилаланина метаболизируется обходными путями, повышается концентрация продуктов его альтернативного метаболизма. Соответственно, в крови и моче повышается их концентрация. Это: фенилпировиноградная кислота, фенилмолочная кислота, фенилацетилглутамин. (Для диагностики используют реакцию Феллинга и тест Гатри.)
Другие продукты этого альтернативного метаболизма: фенилэтиламин, ортофенилуксусная кислота или фенилацетат. Эти соединения практически отсутствуют в норме и являются нейротоксинами, которые нарушают метаболизм липидов в мозге. В сочетании с дефицитом серотонина это ведет к прогрессирующему снижению интеллекта, выявляется через несколько месяцев после рождения. Задержка психомоторного развития столь глубока, что большинство больных детей не ходят (30%) и не говорят (2/3), и лишь в 4-5% слабоумие остается на уровне дебильности. У других – имбецильность или идиотизм.
Основной метод лечения – ограничение потребления фенилаланина с целью снизить его концентрацию в крови с 16мг/дл до 3-12мг/дл. Соблюдение диеты особенно важно до полового созревания и во время беременности (альтернативные метаболиты фенилаланина оказывают тератогенное действие).
Болезнь может быть вызвана и дефектом другого фермента – дигидроптеридинредуктазы. Она не лечится ограничением потребления фенилаланина, имеет худший прогноз.
2 Алкаптонурия – аутосомно-рецессивное заболевание. Причина – дефект оксидазы гомогентизиновой кислоты. Накопление гомогентизиновой кислоты ведет к ее превращению полифенолоксидазой в хиноновые полифенолы. Эти хиноновые полифенолы составляют «охронозный пигмент» (алкаптон), выводимый почками и обусловливающий потемнение мочи больных на воздухе и при подщелачивании. (ПАЦИЕНТЫ В БУКВАЛЬНОМ СМЫСЛЕ ВЫДЕЛЯЮТ ФОТОПРОЯВИТЕЛЬ: при попадании на фотобумагу подщелоченная моча больного вызывает ее почернение!)

Охронозный пигмент экскретируется не полностью и откладывается в соединительно-тканных образованиях: хрящах (делает их хрупкими, что со временем вызывает кальцификацию и артрит), склере (может быть первым проявлением болезни).
Гомогентизиновая кислота (накапливаясь) ингибирует фермент лизилгидроксилазу, участвующую в синтезе коллагена.
Радикально болезнь не лечится. Можно лишь уменьшить степень хондропатии, защищая активность лизилгидроксилазы большими дозами аскорбиновой кислоты.
3 Лейциноз (разветвлено-цепочечная кетонурия) – аутосомно-рецессивное заболевание. При этой патологии нарушено окислительное декарбоксилирование (окисление) разветвленных кетокислот, которые синтезируются путем дезаминирования лейцина, изолейцина, валина.
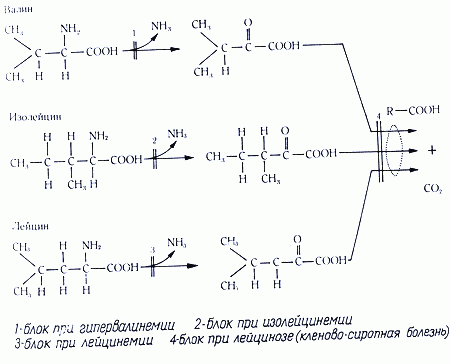
В результате накапливаются и кетокислоты, и вышеперечисленные аминокислоты. Наиболее патогенно накопление лейцина. Лейцин – единственно чисто кетогенная аминокислота, то есть в норме окисляемая до ацетоацетата и ацетилкоэнзима А. Из-за большой роли кетоновых тел в энергообеспечении мозга (особенно при гипогликемии), данная патология чревата развитием умственной отсталости, судорог, мышечной ригидности; также отмечаются гипогликемия, гипотония, больные могут впадать в летаргию, имеется кетоацидоз.
Нарушение обмена лейцина может быть связано с дефектом изовалерил-коА-дегидрогеназы - изовалератацидемия. При обеих болезнях нарушается превращение кетокислот в липиды, => нарушается продукция миелина, страдает энергетика мозга, уменьшается образование ГАМК.
Лечение сводится к специальной диете с ограничением потребления алифатических разветвленных АК. Без лечения – смерть за несколько месяцев.
4 Гомоцистинурия – синдром, имеющий у разных больных разную этиологию. В большинстве случаев причина в дефекте сериндегидратазы, при этом серин и гомоцистеин не могут с должной скоростью образовывать цистатионин. Накапливаются: гомоцистеин, гомоцистин, метионин.
Лечение – витамин В6, активирующий метаболизм гомоцистеина.
Другие дефекты, вызывающие гомоцистинурию – это нарушения в протекании Nметилтетрагидрофолат-гомоцистеин-метилтрансферазной реакции:

· Дефект синтеза апофермента
· Дефицит метилированной формы витамина В12
· Недостаточная активность N-метилентетрагидрофолат-редуктазы
В таких случаях: резистентность к терапии витамином В6; нет накопления метионина; развивается умственная отсталость, эктопия хрусталиков, остеопороз, сколиоз, патологические переломы.
При всех формах гомоцистинурии может быть тромбоэмболический синдром; возрастает риск атеросклероза.
5 Нарушения обмена тирозина:

· Альбинизм. Причина - нарушение образования меланина из тирозина. Наиболее распространенная аутосомно-рецессивная форма (еще м.б. аутосомно-доминантная и сцепленная с полом) основывается на дефекте фермента меланобластов тирозиназы, в норме превращающая тирозин в 3,4-диоксифенилаланин.
Проявления – депигментация волос, сетчатки (вследствие чего родопсин распадается ускоренно => фотофобия), фотодерматит.
· Тирозиноз Медеса. Причина – нарушение активности оксифенилпируватдезоксигеназы. По другим данным, причина в дефиците митохондриальной печеночной тирозинаминотрансферазы. Гомогентизиновая кислота в печени вообще не образуется (в противоположность алкаптонурии).
Развиваются печеночная недостаточность, нефропатия.
· Гипертирозинемия 1 типа. Дефект фумарилацетоацетатгидролазы. Накапливаются тирозин и метионин.
Больные гибнут в младенчестве от печеночной недостаточности (цирроз печени) и нефропатии (тубулонекроз).
2 типа. Дефицит печеночной тирозинаминотрансферазы. Задержка психомоторного развития.
· Хоукинсурия. Дефект 4-гидроксифенилпируватдезоксигеназы. В отличие от остальных миноацидопатий, она аутосомно-доминантная. Тирозинемия.
Задержка психомоторного развития.
· Наследственный первичный гипотиреоз. Дефект йодтирозиндейодиназы. Моно- и дийодтирозин не дейодируются, развивается нехватка тиреоидных гормонов.
Зоб. Нарушение развития мозга и обучаемости.
· Паркинсонизм (см. схемку выше).
96. Нарушения композиции белков плазмы. Значение отдельных белковых фракций плазмы и различных плазменных белков при патологических процессах. Диспротеинемия, гипопротеинемия, гипоальбуминемия, их патологическое значение. Парапротеинемия, виды и этиология. Протеинурия, виды и патофизиологическое значение.
Метод электрофореза на бумаге, с подвижной границей выявляет в плазме здорового человека 5 основных фракций — альбуминовую (54-58%), а также 4 фракции глобулинов — α1 (6-7 %), α2 (8-9 %), β1 (13-14%) и γ (11-12%). Альбумин - основной компонент плазмы, обеспечивающий онкотическое давление (затем α1-глобулины). Факторы комплемента: от α2-фракции (С1s, C9) до γ-глобулиновой (Clq, C8); большинство β1 (СЗ-С5) и β2 (С2, С6, С7). Факторы свёртывания и противосвёртывающей системы в β1-β2-глобулиновой фракции.
Нормальное количество и фракционное распределение - эупротеинемия. Нарушения протеинограммы плазмы – диспротеинемии: ↑ концентраций (гиперпротеинемии), их ↓ (гипопротеинемии) и появление в плазме необычных белков, в норме не присутствующих в значимых количествах (парапротеинемии).
Ложная гиперпротеинемия - при сгущении крови (гиперосмолярной дегидратации). Истинная только при парапротеинемиях, это почти всегда гиперглобулинемия.
Псевдогипопротеинемия - при гемодилюции. Истинные делят на первичные (наследственные) и вторичные (приобретенные). Первичная гипоальбуминемия - у недоношеных (незрелая печень). Наследственная анальбуминемия: альбумины менее 3% белка плазмы, компенсаторно увеличено абсолютное содержание глобулинов. Врождённая агаммаглобулинемия - при Х-сцепленном рецессивном синдроме Брутона (ниже 1 г/л). Гипогаммаглобулинемия - простой вариабельный иммунодефицит. Первичная гипопротеинемия с понижением и альбумина, и глобулинов, особенно γ, - при экссудативной энтеропатии у детей (потеря белка при диарее).
Наиболее важные этиологические факторы вторичных гипопротеинемий:
Пищевая белковая недостаточность (квашиоркор, алиментарный маразм).
Нарушения поступления АК из кишечника при адекватной диете (хронические и рецидивирующие энтериты: целиакия, инфекционные формы спру; наследственные дефекты переваривания и всасывания белков, панкреатическая недостаточность).
Печёночная недостаточность: тормозятся переаминирование и восстановительное аминирование, нарушается балансовая функция в отношении смеси пищевых аминокислот -> гипераминоацидемия, аминоацидурия и дефицит белков печёночного происхождения в плазме: альбумин, трансферрин, протромбин, фибриноген, факторы свёртывания V, VII, IX—XIII. Если болезнь, вызвавшая угнетение функций печени, имеет инфекционную или иммунопатологическую природу – гипербетагаммаглобулинемия (преиммунный и иммунный ответ).
Усиленные внекишечные потери белка через почки. Протеинурия ренальная, преренальная и постренальная. Почечные механизмы протеинурии: повышенная проницаемость клубочкового фильтра и возрастание фильтрации белка, превышающее максимальную производительность системы реабсорбции (острый диффузный гломерулонефрит), понижение реабсорбции белка при неизменной фильтрации (первичный нефротический синдром), увеличение выделения белка эпителием канальцев (синдром Фанкони). При иммунопатологиях – комбинация механизмов. При изолированном нарушении реабсорбции степень селективности высока (наиболее мелкодисперсный белок — альбумин). Если протеинурия неселективна, альбумин в моче до 50%, есть все основные фракции плазменных белков и обычно не менее 10 % - γ-глобулины. Преренальные причины: парапротеинемии. Постренальные причины: гиперсекреция желез мочевыводящих путей и протеинурия, смешанная с пиу- и гематурией (инфекции урогенитального тракта).
Некишечные потери через кожу — плазморея при ожоговой болезни (потеря IgG), эксфолиативный дерматит и синдромы Лайелла и Стивенса-Джонсона + при ↑ гнойных экссудатах (эмпиема), но патогенез затяжных гнойных процессов предусматривает системное действие кахексина и др. цитокинов, вызывающих острофазный ответ, и здесь ограничение синтеза альбумина важнее прямых потерь белка.
Резко увеличенная проницаемость капилляров и венул при шоке, системном действии медиаторов воспаления, системных васкулитах - потеря белков плазмы путём перераспределения между кровью и тканями при недостаточном возврате белка через лимфатические пути. Лимфодинамический отёк при филяриозах («элефантиаз»), конечности не отдают избыток белка из интерстиция в силу окклюзии лимфатических сосудов -> могут превратиться в депо белка, а в крови возможна гипопротеинемия.
При преиммунном ответе TNF, IL-1, 6, 8 вызывают ↑ синтеза гепатоцитами и МФ положительных глобулинов острой фазы, ↓ производства альбумина и трансферрина (отрицательного глобулина острой фазы). Биологический смысл в повышении антиокислительной резистентности тканей, ограничении масштабов альтерации, индукции гипоферремии и гипоцинкемии, что ↓скорость размножения многих бактерий.
Этиология парапротеинемий связана с неопластическими клональными пролиферациями мутантных линий плазмоцитов -> синтез Ig или их отдельных цепей.. При миеломной болезни (Рустицкого-Калера) множественные очаги — колонии миеломных клеток в костях, могут вырабатывать IgG, IgM, IgD, IgA, IgE или свободные лёгкие, либо тяжёлые цепи Ig. Особая форма (только тяжёлые α-цепи Ig) - иммунопролиферативная энтеропатия (средиземноморская лимфома), поражает лимфоидную ткань тонкого кишечника. При плазмоцитоидной лимфоцитарной лимфоме (макроглобулинемия Вальденстрёма) ↑IgM. Ig легко агрегируют на холоде -> холодовой феномен Рейно. При всех парапротеинемиях в моче легкие цепи Ig — белки Бенс-Джонса. Менее значительно количество Ig может возрастать при действии поликлональных иммуностимуляторов.
Криоглобулинемии - нарушения композиции глобулинов плазмы, при которых она имеет тенденцию к формированию желе на холоде (из парапротеинемий встречается только при IgM-секретирующих миеломах и лимфомах).
При патологии из-за деструкции клеток в плазме могут оказываться повышенные количества различных ферментов -> всегда маркёр цитолиза.
97. Нарушения белкового обмена при эндокринных заболеваниях. Патология конечных этапов обмена белка. Гиперазотемия. Креатинурия. Уремия. Гипераммониемия. Этиология, патогенез, виды. Последствия. Роль при возникновении различных видов комы.
Нарушения белкового обмена. Усиление роста свидетельствует об активации синтеза белков или торможении их разрушения. Действительно, введение СТГ животным вызывает задержку азота в организме, положительный азотистый баланс и понижение распада белков. При этом установлено увеличение включения разных аминокислот в белки тканей и снижение отношения остаточного азота к белковому.
Считается, что действие СТГ опосредовано действием пептидных ростовых факторов - инсулиноподобных факторов роста (ИФР), синтезируемых в тканях и прежде всего в печени. Именно с их действием связывают такие анаболические эффекты, как:
1) стимуляция включения SO4 в протеогликаны;
2) стимуляция включения тимидина в ДНК;
3) стимуляция синтеза РНК;
4) стимуляция синтеза белка СТГ. Анаболический эффект СТГ обусловливают два момента:
1. Наличие инсулина. На фоне экспериментального диабета у животных и сахарного диабета у людей СТГ обычно не усиливает синтеза белков. Очевидно, это связано с тем, что инсулин активирует обмен углеводов и стимулирует синтез белка.
2. Концентрация глюкокортикоидов. Малые их дозы способствуют реализации анаболического эффекта СТГ, а большие дозы, наоборот, тормозят анаболический эффект СТГ и задерживают рост, что может быть связано с тем, что кортизол в больших дозах угнетает образование ИФР. У больных с эозинофильной аденомой гипофиза часто усилена продукция глюкокортикоидов. Не исключено, что это один из компенсаторных процессов, направленных на ограничение эффекта избыточных количеств СТГ.
Нарушения белкового обмена при отсутствии инсулина характеризуются преобладанием процессов катаболизма вследствие активации глюконеогенеза из глюкогенных аминокислот и снижения проницаемости клеточных мембран для аминокислот, что приводит к недостатку в тканях свободных аминокислот и нарушению процесса синтеза белка. Стимулируется синтез мочевины, что характеризуется гиперазотемией и приводит к отрицательному азотистому балансу.
Диабетическая кома. Критическая дегидратация тканей организма с поражением
функций головного мозга ведет к развитию диабетической (гипергликемической)
комы. Кома развивается при достижении концентрации глюкозы в крови от 19,4 до 33,3 ммоль/л и более. В этих условиях вследствие кетоацидоза ионы калия выходят во внеклеточное пространство (гиперкалиемия), что лежит в основе нарушения сократительной функции миокарда, а также дыхательной мускулатуры. Диабетическая кома может привести к летальному исходу, если больному не будет своевременно проведена специфическая противокоматозная терапия.
Различают следующие виды диабетической комы:
1. Гипергликемическая кетоацидотическая кома. Развивается чаще всего у больных СД 1 типа вследствие гипергликемии, гиперкетонемии и метаболического ацидоза.
Глюкоза и кетоновые тела выводятся с мочой (глюкозурия и кетонурия), что способствует увеличению осмотического давления в первичной моче, потере ионов Na и сопровождается полиурией. Содержание глюкозы в крови превышает 22 ммоль/л, кетоновых тел - 17 ммоль/л, повышено содержание остаточного азота, мочевины, холестерина, жирных кислот, уровень натрия чаще нормальный, реже - снижен, уровень калия чаще нормальный, у больных с почечной недостаточностью может быть повышен.
2. Гипергликемическая гиперосмолярная кома. Встречается реже, чем кетоацидотическая, и развивается у больных СД 2 типа старше 50 лет при дополнительном воздействии обезвоживающих факторов (рвота, понос, ограничение приема жидкости, ожоги, кровопотеря, полиурия, прием диуретиков). Основными звеньями патогенеза этого вида комы являются дегидратация организма и развитие гиперосмолярности плазмы, уровень гликемии может достигать 55 ммоль/л. У больных нет выраженной гиперкетонемии и кетонурии, отсутствует запах ацетона изо рта и, если не обратиться к врачу, нарастает уровень глюкозы в крови до крайне высокой степени, что способствует усилению диуреза (глюкозурический осмотический диурез). Возникающее обезвоживание приводит к гиповолемии, стимуляции секреции альдостерона и задержке ионов Na и Cl. Показатель осмолярности плазмы повышается в 1,5-2 раза (в норме около 300 мосмоль/л, при коме достигает 500 мосмоль/л), что приводит к резко выраженной внутриклеточной дегидратации, нарушению водного и электролитного равновесия в клетках мозга, гипоксии ЦНС с выраженной неврологической симптоматикой и потере сознания.
3. Гипергликемическая кома с лактат-ацидозом (лактацидотическая). Это относительно редкое, но опасное осложнение СД. В механизме ее развития важную роль играют следующие факторы:
а) снижение активности ферментативного пируватдегидрогеназного комплекса
(выявляется при дефиците инсулина), превращающего пируват в ацетил-КоА. Пируват в обратимой реакции, катализируемой лактатдегдрогеназой, превращается в молочную кислот
б) применение лекарственных препаратов, стимулирующих анаэробный гликолиз и тем самым повышающих содержание лактата и пирувата в организме (например, бигуаниды, повышающие утилизацию глюкозы за счет ее анаэробного распада). При поражении печени или почек может иметь место кумуляция этих препаратов в организме, в результате чего развиваются лактоацидоз и кома;
в) гипоксическое состояние (при котором, как правило, стимулируется гликолиз),
вызванное физическим переутомлением, сердечной или дыхательной недостаточностью.
Гиперкетонемия и кетонурия отсутствуют, могут выявляться незначительная гипергликемия и небольшая глюкозурия. Вследствие несвоевременной диагностики и трудности лечения прогноз может быть неблагоприятным.
4. Гипогликемическая кома. Связана с передозировкой инсулина, препаратов сульфонилмочевины, развитием вторичного гипопитуитаризма (следствие ангиопатии сосудов гипофиза), ослабляющего ответ на гипогликемию, и явлениями диабетического нефросклероза, что удлиняет время циркуляции инсулина и, кроме того, еще более снижает почечный порог для глюкозы, способствуя ее потере.
Причинами гипогликемии могут быть также гиперпродукция инсулина опухолью
поджелудочной железы (инсулиномой), недостаточность контринсулярных гормонов, печеночные формы гликогенозов, заболевания печени, голодание, нарушение расщепления и всасывания углеводов в желудочно-кишечном тракте и др.
В механизме развития гипогликемической комы решающее значение имеет снижение доставки глюкозы к нервным клеткам, что ведет к их энергетическому истощению и нарушению функций ЦНС. При снижении уровня глюкозы менее 3 ммоль/л возникают потливость, тремор, чувство тревоги и голода, слабость. Затем развивается состояние, напоминающее алкогольное опьянение и сопровождающееся дезориентацией, агрессивностью, галлюцинациями. При дальнейшем падении содержания глюкозы (менее 2,5 ммоль/л) возникают клонические судороги и потеря сознания. В тяжелых случаях могут наступать отек и некроз отдельных участков мозга.
Креатинурия в норме характерна только для новорожденных и беременных женщин.
Увеличение выведения креатина с мочой наблюдается
· при мышечной атрофии (наследственные миодистрофии, генерализованной миастении, миозитах)
· голодание
· гипертиреоз
· любые пролонгированные состояния с отрицательным азотистым балансом
Повышение выведения креатина сопровождается уменьшением экскреции креатина и потерю белка.
Уремия - синдром аутоинтоксикации, развивающийся при выраженной почечной недостаточности, в результате задержки в организме азотистых метаболитов и других токсических веществ, расстройства водно-солевого, кислотно-щелочного и осмотического гомеостаза, сопровождающийся вторичными обменными и гормональными нарушениями, общей дистрофией тканей и дисфункцией всех органов и систем.
Ведущую роль в патогенезе уремии, как острой, так и хронической, играет интоксикация продуктами обмена, в норме выводящимися с мочой. Доказано, что в организме больных с уремией аккумулируется большое число органических веществ, особенно продуктов белкового метаболизма, многие из которых обладают токсичностью. Кроме мочевины накапливаются, в частности, аммиак, цианат, креатинин, гуанидины, мочевая кислота, β2-микроглобулин, β2-глюкопротеин, пептиды средней молекулярной массы, аминокислоты, дериваты пиридина, алифатические и ароматические амины, полиамины, индол, фенолы, миоинозитол, маннитол, ацетон, липохромы, циклический АМФ, глюкуроновая и щавелевая кислоты, ряд гормонов, некоторые ферменты и другие.
При ХПН гиперазотемия обусловлена в основном повышением содержания мочевины (ее доля составляет до 80 % от всего небелкового азота), с накоплением которой, помимо диуретического эффекта, связывают появление у больных апатии, тошноты, головной боли и других клинических симптомов. Различная реакция больных на одну и ту же степень гиперазотемии, очевидно, обусловлена двумя факторами: 1) индивидуальной чувствительностью к гиперазотемии. Отдельные больные отмечают плохое самочувствие при умеренной гиперазотемии (концентрация мочевины — 10— 13 ммоль/л). Вместе с тем мы наблюдали девочку 14,5 лет с ХПН вследствие нефронофтиза Фанкони, которая посещала школу при повышении концентрации сывороточной мочевины до 43 ммоль/л; 2) степенью нарушения в этот период других гомеостатических функций почек (ацидоз, водно-электролитные расстройства и др.).
Гипераммониемия — это нарушение обмена веществ, проявляющееся в недостаточности цикла ферментов мочевины, приводящее к отравлению организма аммиаком. Симптомы аммиачного отравления проявляются при превышении этих пределов всего в 2—3 раза. Предельно допустимый уровень аммиака в крови 60 мкмоль/л. При повышении концентрации аммиака (гипераммониемия) до предельных величин может наступить кома и смерть. При хронической гипераммониемии развивается умственная отсталость.
Транзиторной гипераммониемией называется также пограничное состояние, присущее новорожденным детям в период адаптации к внеутробной жизни, проявляющееся обычно на вторые—третьи сутки жизни. Этот вид гипераммониемии встречается чаще всего у недоношенных детей с задержкой внутриутробного развития, с частотой до пятидесяти процентов рождений, однако иногда регистрируется и у доношенных малышей. Часть детей не проявляет симптоматики клинической картины гипераммониемии: признаки угнетения ЦНС (вялость, понижение мышечного тонуса, приступы апноэ, ослабленная реакция зрачков на свет, отказ от еды, ступор и кома), а также расстройства дыхательной функции, желтуха, судороги и обезвоживание. Причиной, вызывающей гипераммониемию, называют кислородное голодание, или гипоксию, во время беременности и в процессе родов.
Приобретённые формы
Приобретённая (вторичная) гипераммониемия развивается вследствие заболеваний печени и вирусных инфекций. В крайне тяжёлых случаях она проявляется как тошнота, рвота, судороги, нечленораздельная речь, затуманивание зрения, тремор, нарушение координации движений.
Наследственные формы гипераммониемии вызваны генетическим дефектом любого из пяти ферментов синтеза мочевины. Соответственно ферменту заболевание делится на пять типов. Первичными признаками гипераммониемий являются сонливость, отказ от пищи, рвота, беспокойство, судороги, нарушение координации движений, тахипноэ, дыхательный алкалоз. Могут развиться печёночная недостаточность, лёгочные и внутричерепные кровоизлияния.
Наиболее частой является гипераммониемия типа II, связанная с недостатком орнитин-карбамоилтрансферазы. Заболевание рецессивно, сцеплено с Х-хромосомой. У матери также наблюдается гипераммониемия и отвращение к белковым продуктам. При полном дефекте фермента наследственные гипераммониемии имеют раннее начало (в период до 48 часов после рождения).
Лабораторным критерием заболевания является накопление глутамина (в 20 и более раз) и аммиака в крови, ликворе и моче.
Причины Гипераммониемии:
Токсичность аммиака обусловлена следующими обстоятельствами:
1. Связывание аммиака при синтезе глутамата вызывает отток α-кетоглутарата из цикла трикарбоновых кислот, при этом понижается образование энергии АТФ и ухудшается деятельность клеток.
2. Ионы аммония NH4+ вызывают защелачивание плазмы крови. При этом повышается сродство гемоглобина к кислороду (эффект Бора), гемоглобин не отдает кислород в капиллярах, в результате наступает гипоксия клеток.
3. Накопление свободного иона NH4+ в цитозоле влияет на мембранный потенциал и работу внутриклеточных ферментов — он конкурирует с ионными насосами для Na+ и K+.
4. Продукт связывания аммиака с глутаминовой кислотой — глутамин — является осмотически активным веществом. Это приводит к задержке воды в клетках и их набуханию, что вызывает отёк тканей. В случае нервной ткани это может вызвать отёк мозга, кому и смерть.
98. Диспротеинозы. Амилоидоз, этиология, патогенез, виды. Роль при патологии. Происхождение и свойства амилоида. Понятие о гиалинозе и роговой дистрофии. Механизмы их формирования.
Диспротеинозы (белковые дистрофии) – процессы накопления в клетках и межклеточном веществе количественно и качественно измененных продуктов обмена белков.
В рез-те гипоксии возникают: мутное набухание, гидропическая дистрофия; вследствие альтерации межклеточного вещества при воспалении и иммунопатологических процессах – мукоидное и фибриноидное набухание). Механизмы мутного набухания (зернистой дистрофии) трактуются как корреляты ранней стадии обратимой гипоксии клетки; механизмы гидропической – как корреляты более глубокой клеточной гипоксии. Мукоидное и фибриноидное набухание – как начальные и глубокие, сопровождаемые коагуляцией, стадии альтерации межклеточного вещества при воспалении. Гиалиново-капельная дистрофия связана с патологией цитоскелета и агрегацией промежуточных филаментов. Также возможно накопление липопротеидов, гликопротеидов, производных аминокислот и т.д.
Амилоидоз – патофизиологический процесс, относительно которого есть явные свидетельства, что в большинстве случаев он связан с определенными расстройствами в иммунном аппарате. Амилоидоз следует рассматривать как группу нарушений, объединяемых по принципу отложения сходно устроенных белковых комплексов. Амилоид – патологический белковый комплекс, который на окрашенным гематоксилином и эозином препаратах выглядит как розовый прозрачный материал, депонированный между клетками в различных тканях и органах. Он обнаруживается при большом числе разнообразных синдромов и болезней.
Все типы амилоида обладают следующими свойствами:
· Окрашивание в красный цвет при окраске йода с последующим изменением цвета на синий или фиолетовый после обработки разбавленной серной кислотой;
· При окраске гематоксилином и эозином в световом микроскопе амилоид выглядит как аморфная, эозинофильная, розовая, прозрачная, внеклеточная субстанция;
· При окраске метиловым фиолетовым – метахромазия;
· Амилоид метится различными антисыворотками против его компонентов при иммуноморфологическом анализе и дает люминесценцию с рибофлавинами S и T.
Различают системный амилоидоз (первичный-связан с иммунодискразиями и вторичный-осложнение основных заболеваний) и местный. Отдельно выделяют наследственный амилоидоз. При первичном наиболее часто вовлекаются почки, средце, печень, селезенка, лимфоузлы, суставы, лучезапястные суставы, кожа, нервы, язык. Вторичный может вовлекать печень, селезенку, почки, сердце, надпочечники.
Имеются два главных элемента амилоидных фибрилл: 90% - фибриллярный компонент, 10% - Р-компонент. Фибриллы белка амилоида образуют типичную уникальную структуру – бета-складчатую листоподобную структуру, не встречающуюся у млекопитающих. Р-компонент, выглядящий как палочка при электронной микроскопии, на разрезе – пятиугольник, составленный из пяти шарообразных структур.
Два типа фибриллярного компонента: AL и AA . Первый формируется при участии плазматических клеток и обычно содержит легкие цепи Ig или иногда их N-концевые участки. Миеломная болезнь (моноклональная злокачественная опухоль), В-клеточные лимфомы осложняются амилоидозом, т.к. ненормальные плазмоциты производят составные части амилоида AL. При образовании злокачественного клона В-клеток происходит то же самое; причем чем злокачественнее клон, тем меньше синтез амилоида и наоборот. Амилоидоз типа AL, произведенным В-клетками, называется первичным (иммунопатологическое расстройство).
Тип АА – этот белок является белком неиммуноглобулиновой природы, предшественники которого синтезируются в печени и, по-видимому, макрофагами. Белок не имеет структурной гомологии с какими-либо белками, сод-ит 76 АК, хар-н для вторичного амилоидоза. Амилоидный предшественник в сыворотке обозначается SAA, циркулирует в составе альфа1-глобулинов в комплексе с ЛПВП третьего подкласса. SAA имеет три формы и принадлежит к положительным глобулинам острофазного ответа. В амилоидогенезе участвуют продукты неполного протеолиза SAA, создаваемые макрофагами, которые захватывают дериваты этого белка в составе иммунных комплексов. Соответственно для обоих типов амилоидоза хар-на связь с длительной активацией иммунной системы и необходима обработка и сборка их компонентов в активных макрофагах.
Кроме предыдущих двух были найдены другие белки в амилоидах:
· Транстиретин (преальбумин) – нормальный сывороточный белок, транспортирующий тироксин и ретинол. Мутантная его форма депонируется при семейной амилоидной нейропатии и семейной амилоидной миокардиодистрофии – аутосомно-доминантные расстройства. Может образовываться без участия фагоцитов.
· Белок бета2-микроглобулин – найден в составе амилоида у пациентов после длительного диализа.
· Бета2-белок амилоида – обнаружен при болезни Альцгеймера. Иногда данный амилоидоз называют старческим мозговым.
Р-компонент идентичен сывороточному альфа1-гликопротеиду, высокогомологичен С-реактивному белку; по-видимому, является острофазным белком, его синтез усиливается при преиммунном ответе. Обеспечивает положительную реакцию с реактивом Шиффа.
Гиалиноз – накопление в соединительной ткани (включая строму и стенки сосудов) любого неамилоидного белкового, гомогенного, плотного, эозинофильного материала. Т.о., одни и те же иммунопатологические процессы могут способствовать депонированию и амилоидного, и неамилоидного белка. Основные механизмы образования: плазматическое пропитывание (гиалин содержит белки плазмы и иммунные комплексы), местная денатурация и коагуляция белков при воспалении и иммунопат. процессах с аутоАТ. Внутриклеточный гиалин при гиалиново-капельной дистрофии и внеклеточный различны. Например, тельца Маллори – гиалиновые включения в гепатоцитах, образовавшиеся в связи с агрегацией белка прекератина, входящего в состав промежуточных филаментов цитоскелета. Тельца Русселя – агрегаты фрагментов АТ в плазм. клетках при интенсивном синтезе Ig. => внутриклеточный гиалин различается по механизму образования.
Роговая дистрофия – избыточное образование кератина при ускорении клеточного цикла и апоптотической гибели кератиноцитов эпидермиса, что может приводить к гиперкератозу и дискератозу (наследственный ихтиоз). Ей способствует гиповитаминоз А. Гиперкератоз характерен также для доброк. опухолей из базальных клеток эпидермиса; образ-ние «раковых жемчужин» - лейкоплакия в карциномах.
99. Нарушения обмена нуклеопротеидов. Причины и механизм нарушений. Гиперурикемия и ее патогенные последствия. Этиология и патогенез подагры. Нарушения обмена пиримидиновых нуклеотидов.

При нарушении катаболических процессов в крови происходит повышение концентрации мочевой к-ты – гиперурикемия.
Повышение уровня мочевой кислоты увеличивает предрасположенность к подагре и почечной недостаточности.
Этиология
Причины первичной гиперурикемии (идиопатическая или врожденная):
1. Около 1% людей, имеющих гиперурикемию, имеют генетический ферментный дефект метаболизма пуринов, который приводит к перепроизводству мочевой кислоты
2. Нарушение различных фаз выведения мочевой кислоты
Причины вторичной гиперурикемии:
1. повышенное образование мочевой кислоты
2. замедление выведения мочевой кислоты
Мочевая кислота — слабокислый продукт, поэтому она, секретируясь в кровь, образует соли-ураты — со щелочными катионами (на 98 % — с натрием), которые связываются глобулинами плазмы только на 5 % и выводятся на 2/3 почками, а на 1/3 — через тонкий кишечник.
Ураты легче выпадают в осадок при температурах ниже 37 °С. Наиболее «холодным» суставом организма является первый предплюснофаланговый сустав стопы.
В моче кристаллизации уратов способствует кислый рН. Именно поэтому все виды ацидоза способствуют обострениям подагры.
Подагра в типичных случаях дебютирует после 40 лет и проходит 4 стадии.
Латентная гиперурикемическая стадия протекает без клинических симптомов и проявляется лишь выявляемой лабораторным анализом гиперурикемией.
Дебютная стадия представляет собой острый моноартрит.
Моноартрит начинается внезапно, на фоне предшествующей гиперурикемии и повышенного содержания уратов в синовиальной жидкости. Подагрический моноартрит характеризуется острейшими признаками воспаления — отеком, краснотой, ограничением функции, повышением местной температуры, а часто — и лихорадкой.
Главным механизмом острого артрита служит реакция сторожевой полисистемы плазмы и полиморфонуклеарных лейкоцитов, в первую очередь — нейтрофильных, на полианионные кристаллы уратов.
1) Ураты активируют классический и альтернативный пути комплемента, фактор Хагемана, а через них — и всю контактную систему полипептидных медиаторов, включая кинины, свёртывание, фибринолиз.
2) Взаимодействие уратовых кристаллов с полиморфонуклеарами и фагоцитами, приводит к освобождению и активации воспалительных медиаторов.
3) Гибель нейтрофилов при фагоцитозе кристаллов ведет к освобождению активных кислородных радикалов, а также особого кристаллозависимого хемотактического фактора, дефензинов и огромного количества лизосомальных гидролаз. Кристаллозависимый хемоаттрактант лейкоцитов, возможно, один из главнейших участников этого «пожара».
4) Макрофаги, фагоцитируя ураты, активируются и выделяют цитокины ИЛ-1, ИЛ-6, ИЛ-8, кахексин, а также простагландины. Это усиливает воспаление и приводит к выделению синовиоцитами коллагеназ, поддерживающих альтерацию.
Самоограничение симптомов артрита зависит от выработки противовоспалительных медиаторов, главным образом, макрофагального и синовиоцитарного происхождения.
Межприступный период подагры характеризуется отсутствием симптомов острого артрита. Несмотря на это сохраняется гиперурикемия, происходит отложение уратов в тканях, прогрессирует нефропатия, и бывают повторные атаки артрита, которые вовлекают тот же и новые суставы. Как и первый приступ, повторные возникают в ответ на тот же разнообразный спектр провоцирующих агентов.
Дата добавления: 2019-03-09; просмотров: 751; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!