На электродах протекают реакции 9 страница
 . (6.18)
. (6.18)
Увеличение давления, которое приложено к раствору, должно быть достаточным для того, чтобы восстановить свободную энергию растворителя в растворе до свободной энергии чистого растворителя. Это увеличение свободной энергии зададим уравнением
dG = VdP при Т = const, (6.19)
так как жидкость несжимаема:
G = VР . (6.20)
Увеличение давления и есть по существу осмотическое давление, т.е. Р = П.
Молярный объем растворителя обозначается символом V. Следовательно, для жидкости  .
.
При равновесии П
 , (6.21)
, (6.21)
откуда
 . (6.22)
. (6.22)
Для разбавленных растворов  .
.
Подставим в уравнение (6.22) и получим:
 . (6.23)
. (6.23)
Поскольку для разбавленных растворов х2 n2/n1, то  ;
;
 – равен объему растворителя и приблизительно объему раствора (для разбавленных растворов). В результате получим:
– равен объему растворителя и приблизительно объему раствора (для разбавленных растворов). В результате получим:
 . (6.24)
. (6.24)
Уравнение (6.24) является специальным случаем уравнения идеального газа применительно к осмотическому давлению. Его можно использовать не только для разбавленных растворов, однако требуется, чтобы пар растворителя вел себя как идеальный газ.
|
|
|
Однако нельзя дать ввести себя в заблуждение видом уравнения и его сходством с уравнением идеального газа, поскольку нет оснований полагать, что осмотическое давление возникает за счет бомбардировки стенок сосуда молекулами растворенного вещества. Из уравнения лишь следует, что растворение одного моля вещества в одном литре растворителя при 0 ºС в идеальном случае будет вызывать повышение осмотического давления на 22,41 атм. Реальные растворы в таких концентрациях дают заметные отклонения от предсказанных осмотических давлений, подобно тому как это имеет место при вычислении точек замерзания и точек кипения.
Уравнение (6.24) можно преобразовать в форму, содержащую молекулярную массу растворенного вещества М. Подставим вместо числа молей растворенного вещества n2 величину g/M, получим:
 или
или 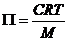 , (6.25)
, (6.25)
где С – концентрация в граммах на л. При делении обеих частей на С слева появляется член П/С, который называется приведенным осмотическим давлением. В идеальных растворах П/С должна оставаться постоянной, т.е. не зависеть от концентрации. Однако это не всегда соблюдается. Общее уравнение для таких случаев имеет вид:
|
|
|
 , (6.26)
, (6.26)
где А1 = 1/МN, а МN – среднестатистический молекулярный вес. Для сильно разбавленных растворов при экспериментально измеряемых концентрациях малыми членами можно пренебречь, и уравнение будет иметь вид:
 . (6.27)
. (6.27)
Такое уравнение правомерно при следующих условиях:
1) величина А2 не очень велика;
2) величина МN не очень велика.
Уравнение (6.27) является уравнением прямой линии с наклоном, численно равным величине А2, называемой «вторым вириальным коэффициентом». Прямая пересекает ось П/CRT в точке, соответствующей величине, обратной MN. Поэтому метод осмотического давления используется для определения молекулярных весов полимеров. Наиболее точные результаты получаются для полимеров с молекулярными весами, лежащими между 10 000 и 20 0000.
6.9. Растворы электролитов
П = iCRT, где i – изотонический коэффициент:
 .
.
Для слабых электролитов i равно отношению фактического числа частиц растворенного вещества к тому числу, которое было бы в отсутствие диссоциации.
Расчет изотонического коэффициента: до диссоциации N частиц, – степень диссоциации, тогда число диссоциированных частиц – N, а недиссоциированных – (1–)N, k – количество образующихся ионов. Тогда при диссоциации образуется N частиц Nk – ионов. Общее число частиц равно:
|
|
|
 .
.
Отсюда изотонический коэффициент:
 или i = 1+(k-1).
или i = 1+(k-1).
Для NaCl Na+ + Cl- k = 2 i = 1 + ;
Для Na2SO4 2 Na+ + SO4- k = 3 i = 1 + 2;
Для Na3PO4 3Na+ + PO43- k = 4 i = 1 + 3 и т.д.
Для сильных электролитов = 1.
Общие сведения о теории
электролитической диссоциации
«В конце концов мы должны признать, что ионная теория неполна, но в химии редко удается достичь совершенства. Наши научные теории совсем не похожи на законы, действующие, подобно сжатым пружинам, сразу же и неумолимо. Наши теории создаются медленно и постепенно, и мы должны честно признать, что ионная теория пока еще переживает пору своей юности. Вместо того, чтобы судить ее по строгим меркам совершенства, давайте просто спросим, чего она уже достигла и что она еще может дать науке», эти слова принадлежат Г. Льюису.
Теория электролитической диссоциации ныне стала одной из незыблемых основ наших научных представлений, и трудно поверить, что докторская диссертация С. Аррениуса, в которой он в 1884 году изложил эту теорию, была принята в университете города Уппсала (Швеция) с наинижайшей из возможных оценок.
|
|
|
В дальнейшем идеи Аррениуса встречали не столько противодействие, сколько полное пренебрежение, но, в конце концов, благодаря объединенным усилиям Я. Вант-Гоффа и В. Оствальда они завоевали всеобщее признание.
Важным вкладом в эту теорию стали последующие работы Ф.Х. Гроттуса, Н.Н. Каяндера и др.
Для обоснования гипотезы электролитической диссоциации имело значение сопоставление: 1) способности разбавленных водных растворов солей, кислот, оснований проводить электрический ток и 2) систематических отклонений некоторых свойств (температуры замерзания, температуры кипения, давления насыщенного пара, осмотического давления и др.) этих растворов и таких же свойств других разбавленных растворов. Была обнаружена количественная взаимосвязь между отклонениями в этих свойствах и способностью проводить электрический ток. Растворы, обнаруживающие большее отклонение в названных свойствах, обладают в общем и большей электропроводностью.
Гипотеза Аррениуса дала возможность объяснить многие особенности в химических свойствах растворов электролитов (реакции гидролиза, значение концентрации водородных ионов и др.). Согласно гипотезе электролитической диссоциации, молекулы солей, кислот и оснований при растворении их в воде претерпевают диссоциацию на ионы.
Однако в гипотезе Аррениуса раствор электролитов рассматривался по существу как механическая смесь из молекул растворителя и ионов и молекул электролита, т.е. в этой гипотезе не находило отражения взаимодействие между всеми этими частицами и поэтому оставалась без объяснения и основная сущность явления.
Развитию гипотезы электролитической диссоциации способствовали работы И.А. Каблукова, В. Нернста, Х. Джонса и др.
Явление электролитической диссоциации наблюдается не только в водных растворах. В других растворителях, особенно обладающих высокой диэлектрической проницаемостью (в том числе и органических), происходит электролитическая диссоциация.
В настоящее время считается, что переход ионов в раствор происходит в результате образования связей между ионом и молекулами растворителя (сольватация) и, в частном случае, молекулами воды (гидратация). Объяснить это можно тем, что в среде с высокой диэлектрической проницаемостью ослабляется сила электростатического притяжения между ионами, равная, согласно закону Кулона,
 .
.
Однако этим дело не ограничивается. Если диссоциация протекает в растворителях, молекулы которых способны к образованию водородных и донорно-акцепторных связей, то этот вид взаимодействия молекул растворителя с частицами электролита может существенно влиять на ход процесса. Так, в растворителях с высокой диссоциирующей способностью (Н2О, НF) именно такое взаимодействие играет часто основную роль в процессе диссоциации. Поэтому в этиловом спирте хлористый водород диссоциирует в сильной степени, а в динитробензоле в очень слабой, хотя диэлектрические проницаемости этих растворителей различаются незначительно.
Работа разъединения ионов при растворении производится главным образом за счет их сольватации (сопровождающейся уменьшением изобарного потенциала системы).
Подобное взаимодействие с молекулами растворителя свойственно не только ионным молекулам, но и молекулам с сильной полярной связью, хотя и в более слабой степени. Это взаимодействие и приводит к диссоциации такой молекулы на ионы. Именно гидратация ионов препятствует обратному соединению ионов в молекулы.
Теплотой гидратации (или энергией гидратации) принято называть количество энергии, отвечающей процессу перевода ионов из газообразного состояния в раствор.
Большое количество энергии, выделяющейся при гидратации (сольватации) ионов, в значительной степени облегчает эндотермический сам по себе процесс диссоциации электролита и вместе с тем стабилизирует ионы, затрудняя обратный процесс их взаимного соединения.
7.2. Сильные и слабые электролиты
По способности к диссоциации электролиты разделяются на сильные и слабые. К сильным электролитам принадлежат сильные кислоты, сильные основания и большая часть солей.
Большинство сильных электролитов кристаллизуются в кристаллах с ионной решеткой, и сильными электролитами часто называют только такие вещества.
К слабым электролитам принадлежат в первую очередь слабые кислоты и слабые основания, а также некоторые соли. Сюда относятся большинство органических кислот, фенолы, амины, угольная и синильная кислота, а также такие соли, как хлорная ртуть, цианистая ртуть и др.
Процесс диссоциации слабых электролитов является обратимым. Для электролита В2А диссоциация представляется уравнением: B2A « 2B+ + A2-.
Как во всяком обратимом процессе, здесь устанавливается равновесие. Количественно его можно характеризовать константой равновесия (константой диссоциации Кд), определяемой для разбавленных растворов того же электролита В2А соотношением:
 , (7.1)
, (7.1)
где СВ+, СА2-, См – концентрации соответствующих ионов и диссоциированных молекул электролита. Соотношение (7.1) применимо к слабым электролитам в разбавленных растворах; в более концентрированных растворах или в присутствии других электролитов в значительной концентрации необходимо пользоваться вместо концентрации активностями. В этом случае соотношение (7.1) приобретает вид:
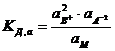 , (7.2)
, (7.2)
где а2В+, аА2- и аМ – активность ионов и недиссоциированных молекул электролита. Напомним, что активностью (точнее термодинамической активностью) данного компонента называется такая величина, которая связана с другими термодинамическими величинами так же, как в идеальных растворах с ними связана концентрация этого компонента.
Коэффициент активности g равен отношению активности к концентрации.
Константу диссоциации, определяемую через активности по уравнению (7.2), называют термодинамической константой диссоциации. Практически, однако, при отсутствии данных об активности часто приходится и для сравнительно концентрированных растворов пользоваться соотношением (7.1).
Рассчитанную таким образом КД называют иногда условной константой диссоциации, поскольку она может несколько изменяться с концентрацией.
Изменение свободной энергии Гиббса в реакциях диссоциации при их выражении через активности выглядит следующим образом:
 . (7.3)
. (7.3)
Оно зависит от состава раствора (концентрации данного электролита и других растворенных веществ раствора). Для стандартных условий
(a'В+ = а'А2- = a'M =1) тогда
 . (7.4)
. (7.4)
Для характеристики состояния слабого электролита в растворе наряду с константой диссоциации пользуются также и величиной степени диссоциации a, которая определяется отношением:
 . (7.5.)
. (7.5.)
Она зависит от следующих факторов:
Ÿ от природы растворителя и растворенного вещества;
Ÿ от температуры (с ростом Т растет и a );
Ÿ от концентрации раствора (с ростом С a уменьшается);
Ÿ от наличия одноименных ионов.
в практике для характеристики силы электролита часто используются величины следующих параметров:
| Параметры | a | Кдисс | рК |
| слабый электролит | < 3% | < 10-4 | >4 |
| средней силы | 3-30% | 10-2-10-4 | 2-4 |
| сильный электролит | > 30% | >10-2 | <2 |
| рК представляет собой – lgKдисс., т.е. рК= – lg Kдисс. | |||
7.3. Закон разведения
В 1888 году немецкий химик В. Оствальд вывел взаимосвязь между a, КД и концентрацией электролита в растворе. Эта взаимосвязь получила название закона разведения. Так, для бинарного электролита (т.е. когда из каждой молекулы образуются два иона)
СВ+ = СА- = a С,
где С – общая концентрация электролита. Тогда
СМ = (1 - a)С,
где СМ – концентрация недиссоциированных молекул.
Подставив эти значения СВ+ ,СА- и СМ в выражение константы диссоциации, получим:
 . (7.6)
. (7.6)
Для очень разбавленных электролитов a мало, и тогда  или
или
 . (7.7)
. (7.7)
V – разведение, т.е. величина, обратная концентрации.
V = 1/С
и тогда
 . (7.8)
. (7.8)
7.4. Сильные электролиты
Уже в работах Д.И. Менделеева, содержащих критику гипотезы электролитической диссоциации, было установлено, что во многих случаях выводы этой гипотезы неприменимы к экспериментальным данным. Опытный материал показывал, в частности, что закон действия масс неприменим к диссоциации сильных электролитов.
Дальнейшее изучение, особенно П. Дебаем и Э. Хюккелем (1923 г.), привело к следующим представлениям: 1) сильные электролиты в растворе полностью диссоциируют; 2) вокруг каждого иона образуется ионная оболочка (ионная атмосфера) за счет противоположно заряженных ионов; 3) молекулы растворителя не только находятся в пространстве между ионами, но и взаимодействуют с ними, образуя сольваты. Это отражается на свойствах как самих ионов, так и молекул растворителя. Конечно, тепловое движение частиц в той или иной мере нарушает указанную закономерность в расположении ионов. Наличие сольватационной оболочки приводит к увеличению радиуса иона и уменьшению скорости его движения, замедлению скорости химической реакции, уменьшению электропроводности раствора.
В растворе всегда экспериментально определяется меньшая концентрация вещества, чем взято для его приготовления. Поэтому различают аналитическую концентрацию (истинную) и активно проявляющуюся концентрацию, или активность: а = fc, где f – характеристика меры электростатического взаимодействия между ионами и молекулами растворителя, названная коэффициентом активности. Для бесконечно разбавленных растворов f = 1; по мере повышения концентрации f сначала уменьшается, а затем растет, преимущественно оставаясь все же меньше единицы.
Неполная диссоциация молекул, взаимное притяжение ионов, их гидратация и другие эффекты влияют на различные свойства раствора. Суммарное влияние их на любое из термодинамических свойств может быть выражено через коэффициент активности электролита в данном растворе. Поэтому коэффициент активности и активность могут быть определены путем измерения различных свойств растворов: температуры замерзания, температуры кипения, давления насыщенного пара, осмотического давления, электродвижущей силы гальванической цепи и др.
7.5. Ионная сила
При рассмотрении термодинамических свойств растворов электролитов широко используется понятие ионной силы. Она определяется как полусумма произведений из концентраций всех ионов в растворе на квадрат их заряда:
 . (7.9)
. (7.9)
Для большинства биологических жидкостей ионная сила равна 0,15. Если в растворе содержатся только однозарядные ионы (т.е. электролит является одно-одновалентным), то ионная сила численно равна просто общему молярному содержанию их в растворе.
Для сильно разбавленных растворов верно следующее правило ионной силы: коэффициент активности f данного электролита в растворе зависит только от ионной силы раствора, и при одинаковом значении ее он сохраняет постоянное значение независимо от вида остальных электролитов, присутствующих в растворе:
Дата добавления: 2018-02-28; просмотров: 316; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!