На электродах протекают реакции 8 страница
или
 . (8.37)
. (8.37)
Из уравнения видно, что при данной температуре константа скорости реакции зависит от константы химического равновесия образования ПК и от частоты распада ПК. Уравнение называется основным уравнением теории переходного состояния.
8.15.4. Термодинамическая форма основного уравнения теории
переходного состояния
Термодинамические данные позволяют связать  со стандартной свободной энергией образования переходного комплекса DG¹, называемой также свободной энергией активации:
со стандартной свободной энергией образования переходного комплекса DG¹, называемой также свободной энергией активации:
DG’¹ = -RT ln , (8.38)
откуда
 . (8.39)
. (8.39)
Тогда для константы скорости химической реакции можно записать
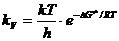 . (8.40)
. (8.40)
Из этого уравнения видно, что скорость реакции определяется свободной энергией активации DG¹, а не Еа, как это следует из теории активных соударений Аррениуса.
Поскольку DG¹ = DН¹ - ТDS¹, то
 (8.41)
(8.41)
или, по теории переходного состояния, kv равна:
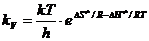 (8.42)
(8.42)
или
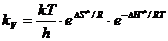 , (8.43)
, (8.43)
т.е. мы получили термодинамическую форму основного уравнения теории переходного состояния.
|
|
|
8.15.5. Сравнение термодинамической формы
основного уравнения теории переходного состояния
с уравнением Аррениуса
При сравнении уравнения для ТПС с уравнением Аррениуса обнаруживается, что
 (8.44)
(8.44)
и соответствует А в уравнении Аррениуса, т.е.
 . (8.45)
. (8.45)
Значит определив опытным путем А, можно рассчитать энтропию активации DS¹, которая дает важную информацию о механизме образования активированного комплекса. Например, чем более жестко связывается фермент с субстратом, т.е. образуется большее число связей, тем больше уменьшается DS¹. Величина DН¹ практически равна энергии активации.
Поскольку А=РZ, то:
 . (8.46)
. (8.46)
Так как Z – теоретическое число столкновений и kT/h практически постоянны, вероятностный фактор Р будет, очевидно, связан с энтропией активации DS¹. Если она велика и положительна, то фактор Р велик, и реакция будет быстрой. Но если DS¹ отрицательна и мала, то реакция будет медленной, т.е. чем больше возрастание энтропии, тем больше вероятность переходного состояния.
|
|
|
Поскольку kT/h соответствует Z и равно при 298К 6,3× 10-12 сек-1, то это число равно числу активных комплексов, разлагающихся за 1 сек. в 1 см3 или количеству соударений в 1 сек. в 1 см3.
Таким образом, скорость реакции, согласно теории переходного состояния, зависит от двух факторов:
1) энергетического фактора – DН¹, энтальпии активации (чем больше энтальпия активации, тем меньше скорость реакции);
2) энтропийного фактора – DS¹, энтропии активации (чем больше энтропия активации, тем больше скорость реакции).
Учет энтропийного фактора для кинетики реакций во многих отношениях оказался плодотворным и впервые позволил установить связь константы скорости со строением молекул реагирующих веществ. При этом теория ПС оперирует, в частности, величинами расстояний между атомами в молекулах, взаимной ориентацией молекул, т.е. параметрами геометрического характера.
Теория активных соударений позволяет при знании энергии активации рассчитать общее число эффективных соударений и отсюда скорость реакции, не объясняя механизма реакции. В отличие от теории активных соударений теория ПК сопоставляет различные возможные комплексы, выявляет большую или меньшую их достижимость и определяет в результате энергетически наиболее выгодный путь реакции.
|
|
|
Для вычисления скоростей взаимодействия двух атомов две теории дают одинаковые результаты. В случае нелинейных многоатомных молекул теория ПК дает значение скоростей, отличных от значений, которые дает теория соударений. Если известна конфигурация реагирующих молекул и активного комплекса, теория ПК позволяет рассчитать предэкспоненциальный множитель. К сожалению, в большинстве случаев строение активированного комплекса и его свойства неизвестны и это затрудняет расчеты.
Таким образом, две теории дополняют друг друга. Теория ПК применяется для вычисления абсолютных скоростей электродных процессов, процессов диффузии и т.д. Теория активных соударений хорошо описывает, как правило, реакции в газовой среде.
Равновесие в гетерогенных системах. Фазовые диаграммы и правило фаз. Уравнение Клаузиса-Клапейрона, уравнения Рауля Коллигативные свойства растворов (температура кипения, температура замерзания, осмотическое давление, давление насыщенного пара) Осмотический потенциал как основа пассивного транспорта
6.5. Уравнение Клаузиуса-Клапейрона
Из рис. 6.1 видно, что наклон ВО и ОА на графике задается производной dp/dT, которая представляет собой скорость изменения давления пара от Т. Наклон ОС численно равен dp/dT – величина, обратная скорости, с которой точка плавления изменяется при изменении давления. Уравнение, которое связывает эти изменения с другими измеряемыми свойствами системы, было предложено в 1834 году Б. Клапейроном и позже модифицировано Р. Клаузиусом.
|
|
|
Это уравнение можно вывести, используя термодинамические соотношения для химического потенциала. В принципе любые свойства (такие, как объем, энтальпия, свободная энергия) зависят от количества вещества и являются экстенсивными. Однако если они относятся к одному молю вещества в строго определенных условиях, они становятся характеристичными свойствами вещества при данных условиях.
Объем, реально занимаемый одним молем растворенного вещества, называется парциальным молярным объемом растворенного вещества. Он соответствует изменению общего объема очень большого количества раствора при добавлении одного моля растворенного вещества. Очевидно, что при добавлении одного моля растворенного вещества концентрация не будет меняться только при условии бесконечно большого объема исходного раствора.
В этом случае для парциального объема можно записать:
 ,
,
где V – общий объем раствора, Т – температура, Р –давление, n – число молей других веществ. Величина V является функцией Т и Р, а также состава раствора.
Если компонент распределен между двумя фазами и , то при равновесии . В случае однокомпонентной системы химический потенциал равен величине свободной энергии, приходящейся на 1 моль: = G и = G.
Если будет слегка меняться при небольших изменениях Т и Р, то аналогичное изменение будет наблюдаться и для (при сохранении равновесия). Поэтому можно записать  .
.
Далее мы знаем, что
dG = Vdp-SdT. (6.4.)
Подставим в это уравнение выражение для парциальных молярных величин
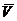 dp - S dT = Vdp - S dT. (6.5)
dp - S dT = Vdp - S dT. (6.5)
Преобразуем выражение (6.5) в виде члена dp/dT
 . (6.6)
. (6.6)
Выражение S по второму началу термодинамики  , где Qобр представляет собой молярную теплоту испарения или молярную теплоту плавления.
, где Qобр представляет собой молярную теплоту испарения или молярную теплоту плавления.
Подставим S в уравнение (6.6) и получим уравнение Клапейрона:
 . (6.7)
. (6.7)
В таком виде это уравнение нельзя проинтегрировать. Р. Клаузиус предложил несколько допущений, что позволяет преобразовывать уравнение в более удобную форму – например, в случае равновесия пар – вода  равно (
равно (  ). Величина Vж для воды составляет приблизительно 0,1% от Vг и поэтому Vж можно не учитывать в члене V(г) – V(ж). Тогда придем к уравнению
). Величина Vж для воды составляет приблизительно 0,1% от Vг и поэтому Vж можно не учитывать в члене V(г) – V(ж). Тогда придем к уравнению
 . (6.8)
. (6.8)
Если принять, что пар подчиняется законам идеального газа, то вместо  запишем RT / P:
запишем RT / P:
 .
.
Заменим выражение для Q более традиционным НV, получим уравнение Клаузиуса-Клапейрона в обычной форме:
 . (6.9)
. (6.9)
Это уравнение можно проинтегрировать, если предположить, что молярная теплота испарения НV является постоянной, т.е. не зависит от температуры. После интегрирования в пределах между Р1, Т1 и Р2, Т2 получаем:

или в форме
 . (6.10)
. (6.10)
6.6. Коллигативные свойства растворов
Рассмотрим, что происходит с растворителем, если в нем растворяют твердое вещество с исчезающе малым давлением пара. Некоторые свойства растворителя могут зависеть лишь от числа добавленных новых частиц. Интересно, что некоторые эти свойства мало зависят от природы растворенного вещества. Это происходит в том случае, если растворенное вещество не взаимодействует с растворителем и не подвергается ассоциации и диссоциации, т.е. число молекул сохраняется постоянным. В таком случае говорят о коллигативных растворах или о коллигативных свойствах растворов.
Четыре свойства из группы коллигативных свойств обычно рассматриваются вместе: понижение давления пара, повышение точки кипения, понижение точки замерзания и осмотическое давление.
Закон Рауля. Точные измерения, которые позволили математически описать влияние растворенного вещества на физические свойства растворителя, были сделаны впервые Ф.-М. Раулем, работавшим в университете Гренобля в середине и конце XIX века (1830-1901 гг.).
Ф.-М. Рауль обнаружил, что давление пара раствора, содержащего нелетучее растворенное вещество, прямо пропорционально концентрации растворителя. Математически это можно записать так:
Р = kx1,
где Р – давление пара раствора, k – константа пропорциональности, х1 – молярная доля растворителя. Когда растворенное вещество отсутствует х1 = 1, то коэффициент k численно равен давлению пара чистого растворителя – Р0. Значит
Р = Р0х1. (6.11)
Поскольку х1+ х2 = 1,0, где х2 – молярная доля растворенного вещества, то мы можем преобразовать уравнение (6.11) в (6.12), подставив вместо х1 = 1 ‑ х2:
 . (6.12)
. (6.12)
Это уравнение показывает, что относительное понижение пара растворителя равно мольной доле растворенного вещества и что давление пара раствора пропорционально молярной доле растворителя. Уравнения (6.11) и (6.12) отражают закон Рауля в преобразованной форме. Закон Рауля и уравнения (6.11) и (6.12) позволяют определить чистоту растворителя и наличие в нем нелетучих веществ.
6.7. Понижение точки замерзания и повышение точки
кипения растворов
Понижение точки замерзания и повышение точки кипения растворов являются прямыми следствиями понижения давления пара.
Рассмотрим двухкомпонентную систему. При растворении одного вещества число химических индивидуальностей возрастает до 2. По правилу фаз
f = c – p + 2 = 2 – p + 2 = 4 – p.
Если существует лишь одна фаза, то у системы должно быть три степени свободы: температура, давление и концентрация.
Рассмотрим вывод формулы для определения повышения точки кипения.
Для этого возьмем уравнение Клаузиуса-Клапейрона поскольку оно является основополагающим соотношением, связывающим давление пара и температуру:
 ,
,
где P0 и Т0 – давление пара и температура кипения чистого растворителя.
Сделаем некоторые упрощения.
а) В соответствии с законом Рауля для идеальных растворов нелетучих веществ P = P0x1.
логарифмирование обеих частей уравнения дает:
lnP = lnP0 + ln x1
или
ln (P0/P) = –ln x1 = –ln (1-x2).
В соответствии с теоремой Маклорена имеем:
–ln(1-x2) = x2 +  x22 + 1/3 x23 + …
x22 + 1/3 x23 + …
Поскольку величина х2 для разбавленных растворов значительно меньше 1, то можно считать, что –ln(1 – х2) х2.
б) Молярная доля растворенного вещества х2 равна n2/(n1 + n2); для разбавленных растворов можно записать, что х2 n2/n1.
в) Поскольку изменение Т происходит в очень узком интервале температур, то Т практически равно Т0. Поэтому вместо Т0Т подставим Т02. Величину Т – Т0 в числителе обозначим Ткип.
Сделав эти упрощения и подставив в уравнение Клаузиуса-Клапейрона, получим:
 . (6.13)
. (6.13)
Для 1000 г воды в качестве растворителя n1 = 1000/18 = 55,6, а n2 – это моляльность раствора. Подставим в это уравнение вместо n2/n1 значение 0,018m. Решая уравнение относительно величины Ткип, т.е. повышения температуры кипения, получаем:
 . (6.14)
. (6.14)
Т кипения воды при давлении 1 атм равна 373,15°. При этой температуре и давлении 1 атм молярная теплота ее испарения равна 18 ´ 539 кал, R = 1,987 кал/мольград. Тогда
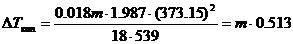 . (6.15)
. (6.15)
Таким образом, мы получили значение эбулиоскопической константы для водных растворов. Аналогично можно получить и соответствующее выражение для криоскопической константы. Размерность этих констант град/моль.
В общем виде уравнение, связывающее изменение температуры кипения или замерзания растворов в сравнении с чистым растворителем, выглядит так:
 , (6.16)
, (6.16)
где К – эбулиоскопическая или криоскопическая константа, m – моляльная концентрация раствора.
6.8. Осмотическое давление
Живым системам присущи различные физико-химические механизмы транспорта растворителя и растворенных веществ. Различают активный и пассивный транспорт. Активный транспорт характерен для живых мембран. Мы пока остановимся на пассивном транспорте, который более изучен и лучше может быть описан математически. Так, в 1748 году А. Ноллет впервые наблюдал, как растворитель проходит через мембрану из разбавленного раствора в более концентрированный. Если к более концентрированному раствору приложить давление, то в зависимости от его величины течение растворителя может быть замедлено, остановлено или обращено.
Осмотическим давлением раствора называется то наименьшее давление, которое, помимо давления самого растворителя, необходимо приложить к раствору, чтобы предотвратить перетекание растворителя к раствору через мембрану, разделяющую раствор и растворитель, причем мембрана непроницаема для молекул растворенного вещества (полупроницаема) в направлении от чистого растворителя к раствору. Растворитель может перетекать из более разбавленного раствора в более концентрированный даже в том случае, если единственной разделяющей поверхностью между двумя растворами будет слой пара.
По-видимому, осмотические эффекты не должны зависеть от природы мембран, используемых для их измерения. В противном случае можно бы было построить вечный двигатель, используя этот эффект.
В 1877 году В. Пфеффер измерил осмотическое давление П нескольких растворов, приготовленных из одного и того же количества вещества в разных объемах растворителя. При этом он показал, что при постоянной температуре произведение П V всегда одно и то же. Он обнаружил также, что для данного раствора с повышением температуры осмотическое давление растет, однако отношение П/Т сохраняется постоянным. Датский химик Я.Вант-Гофф обобщил результаты и предложил эмпирическое уравнение для описания осмотического давления растворов. Это уравнение имеет вид:
П = CRT, (6.17)
где П – осмотическое давление, С – молярная или моляльная концентрация раствора, а Т – температура. Линейная зависимость П от С и Т соблюдается только для идеальных растворов. Таким образом, уравнение Вант-Гоффа служит еще одним примером предельного закона.
Однако уравнение осмотического давления можно однозначно вывести, исходя из теоретических соображений. Осмотическое давление раствора можно интерпретировать как следствие того факта, что давление пара раствора нелетучего вещества ниже, чем давление пара чистого растворителя.
Уравнение осмотического давления легко получить, если предположить, что пар растворителя ведет себя как идеальный газ. Когда раствор отделен от чистого растворителя полупроницаемой мембраной и система путем увеличения общего давления на раствор приведена к равновесию, свободная энергия на моль растворителя должна быть одинакова по обе стороны от мембраны. Уменьшение свободной энергии на моль растворителя в растворе (по сравнению с его свободной энергией в стандартном состоянии) выражается уравнением (6.14):
Дата добавления: 2018-02-28; просмотров: 309; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!