Структурная организация фотосинтетического аппарата
Министерство образования и науки
Донецкой народной республики
Государственное образовательное учреждение
Высшего профессионального образования
«ДОНБАССКАЯ АГРАРНАЯ АКАДЕМИЯ»
Кафедра ЕСТЕСТВЕННОНАУЧНЫХ ДИСЦИПЛИН

Конспект лекций
По учебной дисциплине
«ФИЗИЧЕСКАЯ И КОЛЛОИДНАЯ ХИМИЯ»
Агрохимия и агропочвоведение», 35.03.04 «Агрономия», 35.03.05 «Садоводство» (профиль «Декоративное садоводство и флористика»), 36.03.01 «Ветеринарно-санитарная экспертиза» всех форм обучения
Макеевка, 2020 г.
Министерство образования и науки
Донецкой народной республики
Государственное образовательное учреждение
Высшего профессионального образования
«ДОНБАССКАЯ АГРАРНАЯ АКАДЕМИЯ»
Кафедра ЕСТЕСТВЕННОНАУЧНЫХ ДИСЦИПЛИН
Конспект лекций
По учебной дисциплине
«ФИЗИЧЕСКАЯ И КОЛЛОИДНАЯ ХИМИЯ»
Направления подготовки
Агрохимия и агропочвоведение», 35.03.04 «Агрономия», 35.03.05 «Садоводство» (профиль «Декоративное садоводство и флористика»), 36.03.01 «Ветеринарно-санитарная экспертиза» всех форм обучения
Макеевка, 2020 г.
УДК 544
Чернышева Р. И. Конспект лекций по дисциплине «Физическая и коллоидная химия» для студентов направлений 35.03.03 «Агрохимия и агропочвоведение», 35.03.04 «Агрономия», 35.03.05 «Садоводство» (профиль «Декоративное садоводство и флористика»), 36.03.01 «Ветеринарно-санитарная экспертиза» всех форм обучения/ сост. Чернышева Р. И. - Макеевка: ДОНАГРА, 2020. - 103 с.
|
|
|
Опорный конспект лекций содержит материалы по основным разделам физической и коллоидной химии: основы термодинамики химических процессов, химическая кинетика и катализ, растворы, поверхностные явления, общая характеристика дисперсных систем, строение лиофобных золей, грубодисперсные коллоидные системы, высокомолекулярные соединения и их растворы.
Рецензенты:
Магунова Н.Г., старший преподаватель кафедры естественнонаучных дисциплин;
Шелихов П.В., кандидат биологических наук, доцент, заведующий кафедры естественнонаучных дисциплин.
Рассмотрено на заседании предметно-методической комиссии кафедры естественнонаучных дисциплин
Протокол №__ от “__” ___________ 20___ года
Утверждено на заседании кафедры естественнонаучных дисциплин
Протокол №__ от “__” ___________ 20___ года
Рекомендовано к использованию в учебном процессе Решением Учебно-методического совета ДОНАГРА
Протокол №__ от “__” ___________ 20___ года
© ДОНАГРА, 2020
СОДЕРЖАНИЕ
| ВВЕДЕНИЕ | 5 |
| РАЗДЕЛ 1. ФИЗИЧЕСКАЯ ХИМИЯ | 6 |
| 1.1 Основы термодинамики химических процессов | 7 |
| 1.2 Химическая кинетика и катализ | 17 |
| 1.3 Растворы | 67 |
| 1.4 Поверхностные явления | 76 |
| РАЗДЕЛ 2. КОЛЛОИДНАЯ ХИМИЯ | 80 |
| 2.1 Общая характеристика дисперсных систем | 80 |
| 2.2 Строение лиофобных золей | 83 |
| 2.3 Грубодисперсные коллоидные системы | 92 |
| 2.4 Высокомолекулярные соединения и их растворы | 99 |
| СПИСОК ЛИТЕРАТУРЫ | 102 |
ВВЕДЕНИЕ
|
|
|
Дисциплина «Физическая и коллоидная химия» заканчивает химическую подготовку студентов и является одним из ведущих разделов химической науки, обеспечивая преемственность и связь с неорганической, органической и аналитической химией. Соединение в одном курсе двух научных направлений представляет определенную трудность, так как требует базовых знаний физики и химии.
Раздел «Физическая химия» изучает химическую термодинамику, кинетику и катализ, свойства растворов электролитов и неэлектролитов, поверхностные процессы на границе раздела фаз , то есть все то, что в дальнейшем поможет студентам понять процессы, протекающие в почвах и растениях под действием различных факторов.
Предметом изучения раздела «Коллоидная химия» являются дисперсные системы, их получение, свойства, практическое применение. Для сельскохозяйственных наук (почвоведение, агрохимия) это очень важно, так как почвы, растения и все, что их окружает, являются дисперсными системами. Знание их и умение управлять процессами, в них протекающими, поможет решению ряда теоретических и практических вопросов, поставленных перед наукой и практикой сельскохозяйственного производства.
|
|
|
Раздел 1. Физическая химия
Тема 1.1 Основы термодинамики химических процессов. Предмет химической термодинамики. Основные понятия. Энергия системы, форма обмена энергией с окружающей средой. Энтальпия и энтропия системы. Первое и второе начало термодинамики. Тепловые эффекты химических реакций. Значения химической термодинамики для растениеводства.
Химическая термодинамика исследует закономерности превращений энергии в химических процессах. Она изучает движущие силы химических реакций, их направление и возможности реального осуществления в данных условиях, а также их энергетические характеристики. Химическая термодинамика базируется на трех законах (началах). Законы термодинамики представляют собой обобщение многовекового опыта человечества и являются постулатами.
Термодинамика основана на строгих понятиях: «система», «состояние системы», «функции состояния системы».
|
|
|
Термодинамической системой называют любой объект, состоящий из достаточно большого числа структурных единиц и отделенных от других объектов реальной или воображаемой граничной поверхностью. Объекты природы, не входящие в систему, называют окружающей средой.
Термодинамические системы по характеру обмена веществом и энергией с окружающей средой подразделяют на три типа:
– изолированные – системы не обмениваются с окружающей средой ни массой, ни энергией (Δm = 0, ΔE = 0);
– закрытые – системы обмениваются с окружающей средой энергией, но не обмениваются массой (Δm = 0, ΔE≠ 0);
– открытые – системы обмениваются с окружающей средой и массой, и энергией (Δm ≠0, ΔE ≠0).
Обмен энергией может осуществляться передачей теплоты или совершением работы.
Состояние системы характеризуется термодинамическими параметрами: давлением (р), температурой (Т), концентрацией (С). При изменении параметров меняется состояние системы, осуществляется процесс.
В тех случаях, когда изменение состояния системы происходит при постоянном объеме, процесс называется изохорным (ΔV = 0); при постоянном давлении – изобарным (Δр = 0); при постоянной температуре – изотермическим (ΔТ = 0); при постоянных давлении и температуре – изобарно-изотермическим.
Термодинамические свойства системы – это свойства, изменение которых не зависит от пути процесса. Термодинамические свойства системы можно выразить с помощью нескольких функций состояния, называемых характеристическими. Наиболее используемые характеристические функции: внутренняя энергия (U), энтальпия (H), энтропия (S), энергия Гиббса (G).
Внутренняя энергия (U) включает все виды энергии системы: энергию движения молекул, атомов, ядер и других частиц, а также их потенциальную энергию. Она представляет способность системы совершать работу или передавать тепло. Внутреннюю энергию нельзя измерить, но можно определить ее изменение (ΔU) при переходе из одного состояния в другое: ΔU= U 2 –U 1, где U 2 и U 1 внутренняя энергия системы в конечном и начальном состояниях. Изменение внутренней энергии в процессе можно измерить с помощью работы и теплоты, так как система может обмениваться с окружающей средой энергией в форме теплоты (Q) и работы (W).
Количественные соотношения между изменением внутренней энергии, теплотой и работой устанавливает первый закон (начало) термодинамики:
ΔQ = ΔU +Δ W (1)
ΔQ = ΔU + p ΔV (2)
Теплота, подведенная к системе, расходуется на изменение ее внутренней энергии и на работу системы над окружающей средой. Первый закон термодинамики является следствием выражения закона сохранения энергии, согласно которому энергия не может ни создаваться, ни исчезать, но может превращаться из одной формы в другую.
В тех случаях, когда изменение состояния системы происходит при постоянном объеме (ΔV=0), тогда из математического выражения первого начала термодинамики (2) следует:
ΔQ V = ΔU (3)
Из выражения (3) вытекает термодинамическое определение: внутренняя энергия – функция состояния, увеличение которой равно теплоте QV, полученной системой в изохорном процессе.
Если на систему не действуют никакие другие силы кроме постоянного давления, т.е. при протекании химического процесса единственным видом работы является работа расширения W =p ΔV, то
Q P = ΔU + pΔV (4)
Подставив ΔU = U2 – U1, получим:
QP = U2 – U1 + рV2 – pV1 =(U2 + рV2) – (U1 + pV1) (5)
Сумму (U + pV) называют энтальпией, обозначают Н.
H = U + pV,
QP = Н2 – Н1 = ΔН (6)
Из выражения (6) следует, что энтальпия – функция состояния, увеличение которой равно теплоте, полученной системой в изобарном процессе.
Тепловым эффектом химической реакции называют максимальное количество тепла, которое выделяется или поглощается в необратимом процессе при постоянном объеме или давлении.
Раздел термодинамики, изучающий превращения энергии в химических реакциях, называется термохимией. Уравнение реакции, для которой указываются соответствующие изменения энтальпии, называются термохимическими.
Химические реакции, при протекании которых во внешнюю среду выделяется теплота и происходит уменьшение энтальпии системы (ΔНr<0), называются экзотермическими.
Реакции, в результате которых система поглощает теплоту извне и энтальпия возрастает (ΔНr >0), называются эндотермическими.
Например, окисление глюкозы происходит с выделением большого количества теплоты ΔH = – 2800 кДж, т.е. этот процесс – экзотермический. Соответствующее термохимическое уравнение запишется в виде:
С6Н12О6(к) + 6О2(г) = 6СО2(г) + 6Н2О(г),
ΔНr = – 2800 кДж
В термохимических уравнениях указывают агрегатные состояния веществ: газообразное (г), жидкое (ж), кристаллическое (к), раствор (р).
Для многих реакций изменение энтальпии можно рассчитать с помощью справочных таблиц стандартных энтальпий образования реагентов и продуктов этих реакций. На основе таких расчетов может быть предсказана энергетика без проведения эксперимента.
Энтальпией образования (ΔHf) (formation – образование) соединения называют изменение энтальпии системы, сопровождающее образование 1 моль соединения из простых веществ. Энтальпии образования простых веществ принимают равными нулю. Если исходные вещества и продукты реакции находятся в стандартном состоянии (р = 1 атм = 0,1 М Па, Т = 298 К), энтальпию образования называют стандартной, обозначают ΔHfº, измеряют в кДж/моль; энтальпию реакции в стандартных условиях обозначают ΔHrº.
В 1840 г. Г.И. Гесс сформулировал закон: при постоянных давлении и температуре изменение энтальпии при образовании продуктов из данных реагентов не зависит от числа и вида реакций, в результате которых образуются эти продукты, а определяется начальным и конечным состоянием веществ. Закон Гесса рассматривают как следствие первого начала термодинамики.
В термохимических расчетах чаще применяется следствие из закона Гесса: изменение энтальпии реакции равно алгебраической сумме энтальпий образования стехиометрического количества продуктов за вычетом алгебраической суммы энтальпий образования стехиометрического количества реагентов. Для реакции, представленной в общем виде:
νAA + νBB = νCC + νDD
Следствие из закона Гесса запишется с помощью равенства:
ΔHrº = νC ΔHfº(C) + νD ΔHfº(D) −νB ΔHfº(B) −νA ΔHfº(A)
Энтальпия сгорания (ΔHсº). Для органических веществ не всегда удается определить энтальпию образования, для них определяют энтальпию сгорания. Энтальпией сгорания (ΔHсº) называют тепловой эффект реакции окисления одного моля вещества до высшего оксида в стандартных условиях.
Изменение энтальпии реакции равно алгебраической сумме энтальпий сгорания стехиометрического количества исходных веществ за вычетом алгебраической суммы энтальпий сгорания стехиометрического количества продуктов реакции.
ΔHrº = νA ΔHсº(A) + νB ΔHсº(B) – νC ΔHсº(C) – νD ΔHсº(D)
Многие процессы протекают без подвода энергии, такие процессы называют самопроизвольными. Примерами самопроизвольных химических процессов являются образование ржавчины, взаимодействие щелочных металлов с водой.
Одной из движущих сил химических реакций является уменьшение энтальпии (ΔН < 0). Как показывает опыт, большинство экзотермических реакций протекает самопроизвольно. Однако некоторые эндотермические реакции (ΔН>0) протекают самопроизвольно, например, растворение некоторых солей (КСl, NH4NO3) в воде.
Следовательно, кроме энтальпийного фактора имеется другая движущая сила самопроизвольного процесса. Такой силой является стремление частиц к хаотичному движению, а системы – к переходу от более упорядоченного состояния к менее упорядоченному.
Мерой неупорядоченности состояния системы служит термодинамическая функция, получившая название энтропии.
Состояние системы можно характеризовать микросостояниями составляющих ее частиц, т.е. их мгновенными координатами и скоростями различных видов движения в различных направлениях. Число микросостояний системы называется термодинамической вероятностью системы W. Поскольку число частиц в системе огромно (1 моль содержит 6,02·1023 частиц), то термодинамическая вероятность системы выражается огромными числами. Поэтому пользуются логарифмом термодинамической вероятности lnW. Величина, равная k*lnW = S, называется энтропией системы. Отнесенная к 1 молю вещества, энтропия имеет единицу измерения Дж/моль·К.
Соотношение S = k*lnW установлено Л. Больцманом (1872 г.), где k – постоянная Больцмана, равная отношению газовой постоянной R к постоянной Авогадро k = R/NA = 1,38·10-23Дж/К.
Энтропия есть мера вероятности пребывания системы в данном состоянии или мера неупорядоченности системы.
Важное значение понятия энтропия связано с тем, что на основе этой величины можно прогнозировать направление самопроизвольного протекания процессов. Однако применимость изменения энтропии как критерия направленности процессов ограничивается изолированными системами.
Любой самопроизвольный процесс может протекать в изолированной системе лишь в том случае, когда он характеризуется увеличением энтропии; в равновесии энтропия системы постоянна. Это утверждение, основанное на экспериментальных наблюдениях, является одной из формулировок второго начала термодинамики.
Энтропия вещества в стандартном состоянии называется стандартной энтропией Sº.
В отличие от других термодинамических функций можно определить не только изменение, но и абсолютное значение энтропии. Это вытекает из постулата Планка: при абсолютном нуле энтропия идеального кристалла равна нулю. Этот постулат получил название третьего закона термодинамики.
По мере повышения температуры растет скорость различных видов движения частиц, т.е. число их микросостояний и, соответственно, термодинамическая вероятность и энтропия вещества. При переходе вещества из твердого состояния в жидкое увеличивается неупорядоченность и энтропия. Особенно резко растет энтропия вещества при переходе из жидкого в газообразное состояние, из кристаллического в аморфное.
Энтропия фазовых переходов при р=const ΔS = ΔH/Т. Энтропия является функцией состояния системы. Изменение энтропии системы в результате протекания реакции (ΔSr) равно сумме энтропий продуктов реакции за вычетом энтропий исходных веществ с учетом стехиометрических коэффициентов. Например, изменение энтропии реакции (ΔSrº):
СН4(г) + Н2О(г) = СО(г) + 3Н2(г) ΔSrº =
= Sº(СО) + 3 Sº(Н2) – Sº(Н2О) – Sº(СН4)
Пользуясь справочными данными находим Sº веществ.
ΔSrº = 197,54 + 3·130,58 – 186,19 – 188,7 = 219,39 Дж/К
Энтропия системы в результате реакции возросла, это говорит о переходе системы от более упорядоченного состояния к менее упорядоченному.
В химических процессах проявляются две тенденции. С одной стороны, стремление к образованию прочных связей между частицами, сопровождающееся понижением энергии системы. Эта тенденция характеризуется в изобарно- изотермических условиях энтальпийным фактором и количественно выражается через ΔН. Вторая тенденция проявляется в стремлении к разъединению частиц, к беспорядку, она характеризуется энтропийным фактором и количественно выражается произведением абсолютной температуры на энтропию процесса (Т*ΔS).
Энтальпийный и энтропийный факторы, характеризующие две противоположные тенденции процессов, взятые по отдельности, не могут быть критериями самопроизвольного течения химических реакций. Для изобарно- изотермических процессов их объединяет функция, называемая энергией Гиббса процесса:
ΔG = ΔH – TΔS
Энергия Гиббса служит критерием самопроизвольного протекания химической реакции при изобарно- изотермических процессах. Химическая реакция принципиально возможна, если энергия Гиббса уменьшается, то есть ΔG<0. Это уравнение является условием возможности самопроизвольного протекания реакции в прямом направлении. Химическая реакция не может протекать самопроизвольно, если энергия Гиббса возрастает, то есть ΔG>0. Термодинамическим условием возможности самопроизвольного протекания обратной реакции является возрастание энергии Гиббса ΔG>0. Если ΔG=0, то реакция может протекать как в прямом, так и в обратном направлениях, то есть реакция обратима.
Направление химических реакций зависит от их характера. Условие ΔG<0 соблюдается при любой температуре для экзотермических реакций (ΔН<0), у которых ΔS>0. У таких реакций обе движущие силы (ΔН и ТΔS) направлены в сторону протекания прямой реакции и ΔG<0 при любых температурах. Такие реакции самопроизвольно могут идти только в прямом направлении, то есть являются необратимыми. Примером такой реакции служит реакция:
С(графит) +1/2 О2 = СО
Эта реакция экзотермична (ΔН= –110,5 кДж/моль), в результате ее протекания возрастает число молей газообразных веществ (ΔS=89,38 Дж/моль·К), то есть при любых температурах ΔG<0.
Эндотермическая реакция (ΔН>0), в результате которой уменьшается число молей газообразных веществ (ΔS<0), не может протекать самопроизвольно в прямом направлении при любой температуре, так как всегда ΔG>0. Возможности протекания многих реакций зависят от температуры, так как с изменением температуры меняется знак энергии Гиббса этих реакций. Если в результате экзотермической реакции ΔН<0 и ΔS<0, то при невысокой температуре |ΔН| < |ТΔS|, и реакция может самопроизвольно протекать в прямом направлении (ΔG<0). При высоких температурах |ΔН| > |ТΔS|, и прямая реакция самопроизвольно протекать не может (ΔG>0), а обратная возможна.
Энергия Гиббса является функцией состояния процесса. Изменение энергии Гиббса системы при образовании 1 моля соединения из простых веществ, устойчивых при 298 К, называется энергией Гиббса образования. Если вещество находится в стандартном состоянии, то энергия Гиббса называется стандартной ΔGfº. Единица измерения: кДж/моль. Энергия Гиббса является функцией состояния, то есть ее изменение не зависит от пути протекания процесса, а лишь от исходного и конечного состояний системы. Поэтому энергию Гиббса химической реакции ΔGr (кДж) можно рассчитать как сумму энергий Гиббса продуктов за вычетом энергий Гиббса исходных веществ с учетом стехиометрических коэффициентов.
Например, изменение энергии Гиббса получения водорода методом взаимодействия метана и водяного пара:
СН4 (г) + 2Н2О (г) = СО2(г) + 4Н2(г)
рассчитывается по уравнению:
ΔGºr = ΔGºf (СО2) + 4ΔGºf (Н2) – ΔGºf (СН4) – 2ΔGºf (Н2Ог)
ΔGfº простых веществ принимают равными нулю.
Формирование урожая и качество растениеводческой продукции непосредственно связаны не только с обменом веществ, но и с обменом энергии, необходимой для осуществления всех жизненно важных процессов, протекающих в растениях. Применение законов химической термодинамики к биохимическим реакциям позволяет установить, будет ли данный процесс протекать самопроизвольно или он потребует подвода энергии извне; сколько энергии может выделиться, или использоваться растением в результате этого процесса, какова вероятность его осуществления в данных условиях. И в целом растении, и в каждой его клетке энергия не возникает из ничего и не исчезает бесследно, все взаимные переходы различных форм энергии происходят в строго эквивалентных количествах. Самопроизвольно идут те биохимические процессы, которые сопровождаются уменьшением энергии Гиббса. Это ферментативное расщепление крупных молекул белков, углеводов, жиров, в результате которых образуются менее сложные органические соединения (так называемый катаболизм). Энергия, выделяющаяся в ходе этих процессов, а также световая энергия, получаемая растением в ходе фотосинтеза, используется для реакций, сопровождающихся усложнением молекул (анаболизм). Процессы расщепления и синтеза взаимосвязаны, сопряжены друг с другом.
В растениях (открытых системах) в процессе жизнедеятельности энтропия может как увеличиваться, так и уменьшаться, однако энтропия системы растение – окружающая среда в целом растет. Растет энтропия в процессе гибели организма, когда синтетические биохимические реакции прекращаются.
Однако в живой природе изменение энтропии не является движущей силой эволюционного процесса, который подчиняется не термодинамическим, а биологическим закономерностям.
Практически все химические процессы протекают с изменением количества тепла в системе. Одним из экспериментальных методов изучения тепловых эффектов реакций является калориметрия.
Рассмотрим калориметрические измерения тепловых эффектов процесса растворения соли и реакции нейтрализации.
Растворение является сложным физико-химическим процессом. При растворении твердого вещества происходит разрушение его кристаллической решетки, для чего требуется подвод энергии, следовательно, происходит поглощение тепла, процесс эндотермический (ΔHºr > 0). Молекулы соли распадаются на ионы, которые взаимодействуют с растворителем, происходит процесс сольватации, если растворителем является вода – гидратации. Сольватация и гидратация сопровождаются выделением тепла, то есть являются экзотермическими процессами (ΔHºr < 0). В связи с этим процесс растворения твердого вещества характеризуется тепловым эффектом, который рассчитывается как сумма:
ΔH М = ΔH реш + ΔH гидр,
где ΔH М – энтальпия растворения 1 моля вещества, Дж/моль;
ΔH реш – энтальпия разрушения кристаллической решетки, Дж/моль;
ΔH гидр – энтальпия гидратации, Дж/моль.
В зависимости от соотношения значений теплоты процесс растворения может быть экзотермическим (ΔHº<0) или эндотермическим (ΔHº>0). Для процесса растворения большое значение имеет количество взятого растворителя. Увеличение объема растворителя приводит к такому состоянию, когда дальнейшее прибавление не влияет на величину теплового эффекта, т. е. теплота растворения будет постоянной величиной.
Молярной энтальпией растворения твердого вещества называют тепловой эффект, сопровождающий процесс растворения 1 моля вещества в таком количестве растворителя, дальнейшее добавление которого не влияет на величину теплового эффекта.
Нейтрализацией называют реакцию взаимодействия кислоты и основания. Реакция нейтрализации разбавленного водного раствора 1 моля любой сильной кислоты любым сильным основанием всегда сопровождается одинаковым экзотермическим эффектом:
ΔHºr = –57,3 кДж/моль
Эта величина является тепловым эффектом реакции образования 1 моля Н2О из ионов: Н+ + ОН– = Н2О.
Тепловой эффект реакции нейтрализации 1 моля разбавленного раствора слабой кислоты сильным основанием или слабого основания сильной кислотой не будет постоянной величиной, т. к. на его величину оказывает влияние процесс диссоциации слабой кислоты или слабого основания.
Калориметрические исследования связаны с определением теплоемкости калориметрической системы. Теплоемкость системы (символ С, единица измерения Дж/град.) – количество теплоты, которое необходимо подвести к системе, чтобы повысить температуру на один градус. Теплоемкость тела рассчитывается по формуле С = Q/Δt. Удельная теплоемкость (символ с, единица измерения Дж/кг·град.) рассчитывается по формуле с = С/m, где m – масса тела.
Все калориметрические измерения можно проводить в упрощенной калориметрической установке (рис.1).
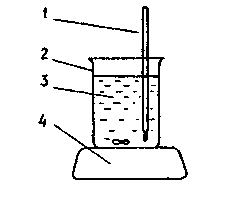
Рисунок 1.1 - Установка для калориметрических исследований: 1 – термометр; 2 – калориметрический стакан;3 – раствор соли;4 – магнитная мешалка
Тема 1.2 Химическая кинетика и катализ. Порядок реакции и константы скорости реакции. Необратимые реакции. Виды катализа. Энергия активации химической реакции . Фотохимические процессы. Фотосинтез.
Изучение термодинамических характеристик химических реакций позволяет определить возможность и степень их протекания, рассчитать условия нахождения системы в состоянии равновесия и т.п. Однако, на ряду со многими достоинствами термодинамического подхода, он не дает ответа на вопрос о скорости химического процесса и механизме его протекания, так как в термодинамических уравнениях отсутствуют переменные, зависящие от времени, а вместо конкретного пути реакции рассматриваются только конечное и начальное состояния системы.
Рассмотрение вопросов, связанных с механизмом и закономерностями протекания химических реакций во времени, является задачей химической кинетики. Химическая кинетика – учение о закономерностях протекания химического процесса во времени и его механизме. Под механизмом реакции понимается совокупность всех элементарных стадий реакции и их конкретное содержание, то есть по какому пути протекает процесс, каковы его промежуточные продукты и что происходит при взаимодействии частиц. Например, для взаимодействия газообразных хлора и водорода можно представить, что вначале молекула хлора распадается на атомы, затем один из атомов хлора взаимодействует с молекулой водорода и т.д.:
Сl2→2Cl·
Cl· +H2→HCl+H·
H· + Cl2→HCl +Cl·
H +H·→H2
Cl·+Cl·→Cl2
точками здесь обозначены неспаренные электроны атомов. Такая схема иллюстрирует механизм реакции с точки зрения пути протекания. Механизм отдельных элементарных стадий показывает, что происходит с атомом хлора и молекулой водорода при их взаимодействии. Элементарная стадия реакции является совокупностью множества элементарных актов, каждый из которых приводит к изменению химического строения реагентов. К каждому элементарному акту реакции применимо понятие молекулярности: Молекулярность равна числу частиц, взаимодействующих одновременно в элементарном акте реакции. Поскольку вероятность одновременной встречи четырех и более активных частиц крайне мала, то существуют только моно-, би- и тримолекулярные реакции, причем даже тримолекулярные реакции очень редки.
В качестве скорости химической реакции рассматривают величину изменения количества вещества за единицу времени в единице объема или изменение концентрации реагирующего вещества за единицу времени, если объем системы остается постоянным. Скорость реакции можно определять либо по убыли концентрации одного из реагентов, либо по приросту концентрации одного из продуктов реакции. Так как по определению скорость реакции – величина положительная, то в общем случае можно записать:

где знак плюс берется при рассмотрении концентрации продукта реакции, минус – при рассмотрении концентрации реагента. Поскольку стехиометрические коэффициенты веществ, участвующих в реакции, в общем случае различаются, то и скорости, определенные через изменение концентрации различных веществ, будут разными. Поэтому на практике используют понятие истинной скорости, равной отношению скорости, определенной по изменению какого-либо участника реакции, к стехиометрическому коэффициенту νi данного вещества:

Если использовать величину химической переменной χ, которая определяется соотношениями

то для химической реакции, записанной в общем виде

где Ai – реагенты, Bj – продукты реакции, ai и bj – стехиометрические коэффициенты, истинная скорость определяется уравнением
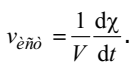
Согласно основному постулату химической кинетики, скорость реакции пропорциональна концентрациям реагирующих веществ, взятых в некоторых степенях. Это положение еще иногда называют законом действующих масс, а уравнение, его описывающие – кинетическим уравнением реакции. Так, для реакции, записанной выше в общем виде, скорость будет равна

где k – константа скорости реакции (удельная скорость), равная скорости реакции при единичных концентрациях реагирующих веществ, n1, n2, ni – порядок реакции по данному веществу. Сумма порядков по всем веществам определяет общий порядок реакции. Константа скорости реакции зависит от температуры и природы реагирующих веществ. Константы скорости для реакций разного порядка имеют различные единицы измерения, что позволяет сопоставлять их только для реакций с одинаковым порядком. Для реакций различного порядка сравнивать можно только скорости. Порядок реакции по данному веществу в общем случае не равен стехиометрическому коэффициенту данного вещества и может быть нулевым или дробным. Объясняется это тем, что большинство реакций протекают по сложному механизму через ряд промежуточных стадий, а химическое уравнение отражает только материальный баланс совокупности элементарных стадий. Кроме того, экспериментально определяемый порядок реакции в зависимости от концентрации реагентов может изменять свое значение. Совпадение стехиометрического коэффициента и порядка наблюдается только для элементарных стадий реакции. В отличие от молекулярности, которая имеет четкий физический смысл, порядок реакции является формальной величиной.
В экспериментальной практике применяют различные способы наблюдения за развитием химической реакции во времени. Удобными для проведения кинетического исследования являются реакции, протекание которых может быть приостановлено в определенный момент времени, добавлением, например, какого-либо реагента, тормозящего процесс или охлаждением системы. Остановив протекание реакции в такой системе, производят затем химический анализ ее состава. Наиболее удобны для проведения кинетического исследования методы, основанные на непрерывном наблюдении за состоянием системы. Выбор подходящего метода зависит прежде всего от природы реагирующих веществ и физико- химических свойств системы в целом. Особенно ценными являются такие методы, которые допускают непрерывное наблюдение за ходом реакции без какого-либо вмешательства в ход реакции, как, например, измерение вращения плоскости поляризации и светопоглощения. При исследовании газовых реакций, протекающих с изменением числа молекул, кинетические расчеты легко проводить на основе наблюдений за изменением общего давления в системе. При изучении реакций в растворах, которые сопровождаются выделением малорастворимых газов, например, азота или кислорода, расчеты основываются на измерении давления выделяющегося газа. Для наблюдения за реакциями, которые сопровождаются изменением концентрации кислот, щелочей, галогенидов, применяют периодический отбор проб для титрования. При изучении кинетики реакций в растворах исследование проводят на основе таких физических свойств, как светопоглощение, вращение плоскости поляризации, электропроводность, коэффициент преломления.
При кинетическом исследовании химического процесса вначале экспериментально определяют скорость реакции. Затем находят порядок ре акции по каждому реагенту и константу скорости. Затем эти данные анализируются с целью выяснения механизма реакции, соответствующего установленному кинетическому уравнению.
Наиболее простыми для кинетического исследования являются необратимые реакции, то есть такие, в которых равновесие сильно смещено в сторону образования продуктов реакции. Это происходит, например, при выходе одного из продуктов из сферы реакции в виде газа или осадка.
Самое простое кинетическое уравнение имеют реакции нулевого порядка. Такой порядок наблюдается, например, для реакции, протекающей на границе раздела фаз, когда убыль реагентов вблизи поверхности постоянно восполняется их диффузией из глубины фазы. Для таких реакций скорость является величиной постоянной
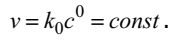
Рассмотрим необратимую реакцию первого порядка, которая в простейшем случае соответствует записи
А → продукты.
Такому уравнению отвечает, например, реакция разложения паров диметилового эфира
СН3ОСН3 → СН4 + Н2 + СО.
Если начальная концентрация вещества А равнялась сo,A , а через некоторое время она уменьшилась на x , то скорость реакции в любой момент времени можно рассчитать по уравнению

Отсюда, интегрируя получаем
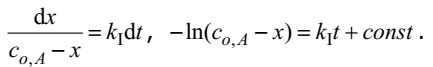
Константа интегрирования может быть определена из условия, что в момент времени t = 0, то есть до начала реакции, x = 0, поэтому ,
const c = − ln сo,A ,
и ,

где cА= co,A-x концентрация вещества А в момент времени t . Эта концентрация может быть рассчитана по уравнению
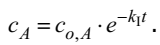
Важным параметром, используемым при кинетическом описании химического процесса, является время полураспада (период полупревращения) – время, в течение которого прореагирует половина исходного вещества. Очевидно, что для реакции первого порядка

Согласно этим уравнениям, для реакции первого порядка концентрация реагента убывает по экспоненциальному закону, линия в координатах
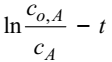
является прямой, а период полупревращения не зависит от начальной концентрации реагента. Кинетическим уравнением первого порядка описывается реакция инверсии сахарозы с образованием глюкозы и фруктозы

Несмотря на то, что по своему механизму эта реакция является бимолекулярной, для нее характерны кинетические закономерности реакций первого порядка. Согласно основному постулату химической кинетики, скорость этой реакции определяется выражением

Так как концентрация воды значительно больше концентрации сахарозы, то величина cH2O в ходе реакции остается практически постоянной. Поэтому для скорости реакции можно записать
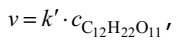
где k=k’*cH2O=const. Реакции такого типа называют псевдомономолекулярными.
В качестве другого примера несоответствия порядка реакции виду химического уравнения можно привести реакцию разложения азотного ангидрида
2N2O5 → 4NO2 + O2.
Наиболее вероятным представляется протекание этой реакции через следующие стадии
N2O5 → N2O3 + O2,
N2O5 + N2O3 → 4NO2.
Первая стадия является мономолекулярной и протекает медленно, а вторая – бимолекулярной и быстрой. Так как скорость последовательной реакции определяется наиболее медленной стадией, то в целом кинетика реакции разложения азотного ангидрида будет соответствовать первому порядку. Рассмотрим необратимую реакцию второго порядка. В качестве примера реакции такого типа можно привести реакцию омыления сложного эфира щелочью
СН3СООС2Н5 + NaOH → CH3COONa + C2H5OH.
Если начальные концентрации обоих веществ одинаковы и равны co, а через некоторое время они уменьшились на x, то скорость реакции в любой момент времени можно рассчитать по уравнению

Откуда, разделяя переменные и интегрируя, получаем уравнение
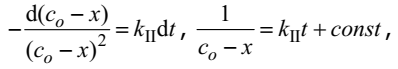
в котором константа интегрирования также как и в предыдущем случае может быть определена из начальных условий; const=1/c0. Вводя обозначение для текущей концентрации реагентов c=с-x , имеем


Таким образом, для реакции второго порядка концентрация реагента убывает, линия в координатах 1/c – t является прямой, а время полураспада обратно пропорционально начальной концентрации реагентов. Совершенно аналогично для простейшего случая необратимой реакции третьего порядка при единичных стехиометрических коэффициентах и равных начальных концентрациях реагентов, получим




Таким образом, для реакции третьего порядка концентрация реагента убывает, прямой линией является график в координатах 1/c2 – t , а период полураспада обратно пропорционален квадрату начальной концентрации.
Теория активных соударений для объяснения кинетических закономерностей реакций использует основные положения молекулярно-кинетической теории о движении молекул газов и понятие об энергетическом барьере, который необходимо преодолеть реагентам для образования продуктов реакции. Основное предположение теории заключается в том, что для протекания реакции необходимо столкновение молекул реагентов. Поэтому константа скорости реакции пропорциональна числу соударений молекул в единице объема системы. Согласно молекулярно-кинетической теории газов, число соударений молекул А и В в единице объема определяется выражением

где cA , cB – количество молекул реагентов в единице объема; dAB – эффективный диаметр столкновения – расстояние между центрами молекул, при котором происходит акт химического взаимодействия, в простейшем случае эта величина равна сумме радиусов реагирующих молекул dAB=rA+rB; MA и MB – молярные массы реагентов. Величина zo очень велика. Если бы каждое столкновение молекул приводило к реакции, то практически все они протекали бы мгновенно. Однако для протекания реакции необходимо выполнение еще ряда условий:
– молекулы должны обладать повышенной энергией по сравнению с обычным состоянием; число столкновений молекул, обладающих достаточной для протекания реакции энергией E , согласно распределению молекул по энергии Больцмана, равно

– кроме наличия достаточной энергии при столкновении молекулы должны быть так ориентированы в пространстве, чтобы их реакционные центры пришли во взаимодействие, это учитывается введением множителя P, называемого стерическим фактором.
Таким образом, скорость бимолекулярной реакции определяется выражением

Так как, согласно основному постулату химической кинетики

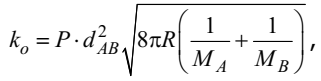
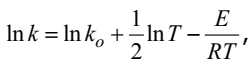

В соответствии с уравнением Аррениуса

поэтому
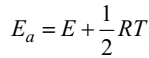
Таким образом, энергия активации определяется в основном избыточной энергией молекул. Необходимость наличия избыточной энергии молекул для активации можно объяснить следующими соображениями. Рассмотрим бимолекулярную реакцию
АВ + СD → АС + ВD.
Прежде чем произойдет превращение молекул АВ и CD в молекулы АС и BD, они должны сблизиться на такое расстояние, когда возможно перераспределение электронной плотности между атомами, входящими в состав реагентов. Очевидно, что это требует затраты энергии на преодоление сил отталкивания. Для перераспределения электронной плотности с образованием молекул продуктов также требуется некоторая энергия для разрыва или частичного ослабления исходных химических связей в молекулах АВ и CD. Таким образом, для протекания реакции необходимо сообщить реагентам некоторую избыточную энергию. Эта энергия обычно значительно меньше энергии связей в реагирующих молекулах, так как присутствие молекулы другого реагента способствует перераспределению электронной плотности в молекулах. Описанный процесс взаимодействия молекул изображен на схематическом рисунке 1.2, на ординате которого отложена энергия реагирующей системы U на различных этапах протекания реакции.

Рисунок 1.2 - Путь химической реакции
В начальном положении обе молекулы АВ и CD находятся на достаточно большом расстоянии одна от другой и система имеет начальную энергию U0. При сближении реагирующих молекул начинается перераспределение связей, на что требуется затратить некоторую энергию, равную энергии активации. После произошедшего химического превращения молекулы AC и BD расходятся на достаточно большом расстояние и конечная система приобретает энергию U1. Разность величин U1 и U0 соответствует тепловому эффекту реакции. Несмотря на то, что в целом при образовании молекул АС и BD энергия выделяется (∆<H0), реакция все же не идет до тех пор, пока не произойдет увеличения энергии исходной системы до значения, отвечающего вершине энергетического барьера. Высота этого барьера и равна энергии активации Ea.
Катализом называют явление изменения скорости химической реакции в присутствии веществ, которые не входят в стехиометрическое уравнение химической реакции, а их количество и состав до и после реакции остается неизменным.
Вещества, изменяющие скорость реакции, но не расходующиеся в результате ее протекания, называют катализаторами. Количество катализатора может быть очень небольшим, но скорость реакции в первом приближении пропорциональна количеству катализатора. В некоторых реакциях катализатором может быть один из продуктов, такие реакции называют автокаталитическими.
Скорость реакции может возрастать под действием катализатора, в этом случае катализ называют положительным, при уменьшении скорости реакции в присутствии катализатора – отрицательным.
Для обратимой реакции катализатор не смещает положения равновесия, а только ускоряет его достижение. Это и понятно, так как начальное и конечное состояния системы с катализатором и без него одинаковы.
Действие катализатора специфично, то есть любой катализатор оказывает влияние только на определенную группу реакций. Например, медь и никель наилучшим образом подходят для проведения реакций гидрирования и дегидрирования, серебро – для реакций окисления. Особенно сильно специфичность выражена у ферментов – природных катализаторов биологических процессов.
Активность катализатора может быть изменена в результате воздействия некоторых веществ, которые сами катализаторами не являются. Если эти вещества усиливают действие катализатора, их называют промоторами, если снижают – каталитическими ядами или ингибиторами. Например, как катализатор при синтезе аммиака используется железо, промотированное оксидами щелочных металлов. В качестве каталитических ядов чаще всего выступают соединения серы, мышьяка, свинца и др. Различают гомогенный катализ, когда и катализатор, и реагирующие вещества находятся в одной фазе, и гетерогенный, или контактный, катализ, при котором катализатором обычно является твердое вещество в газообразной или жидкой реакционной смеси. Влияние катализатора на скорость реакции объясняется тем, что он входит в состав активного комплекса и изменяет энергию активации реакции. При распаде активного комплекса происходит регенерация катализатора, при этом количество катализатора остается постоянным, но его физическое состояние может измениться. Схематически для реакции типа

путь протекания в присутствии катализатора можно представить так
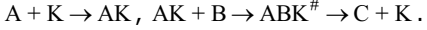
Например, при получении серной кислоты для окисления диоксида серы до триоксида в качестве катализатора используют NO. Суммарная реакция
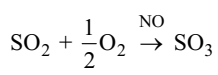
может быть представлена как сумма ниже приведенных стадий
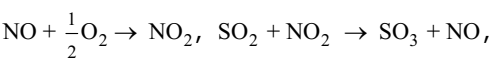
в которых в качестве промежуточного соединения катализатора с реагентами выступает диоксид азота.
Схематически изменение потенциальной энергии системы при протекании реакции представлено на рисунке 1.3. Изменение величины энергии активации ∆Ea соответствует изменению константы скорости реакции по сравнению с обычными условиями протекания в соответствии с уравнением
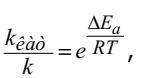
где kêàò – константа скорости каталитической реакции.

Рисунок 1.3 - Путь каталитической и некаталитической реакции
При гетерогенном катализе большое значение имеют стадии адсорбции реагентов на поверхности твердого катализатора и последующей десорбции продуктов. В существующих ныне теориях гетерогенного катализа предполагается, что в результате адсорбции реагентов образуются либо неустойчивые промежуточные продукты взаимодействия с катализатором, либо повышается их химическая активность, что и приводит к снижению энергии активации. Адсорбция реагентов происходит на активных центрах поверхности катализатора, в качестве которых выступают места ненасыщенности силового поля кристаллической решетки катализатора. Ими могут быть ребра, трещины, выступы и другие места неоднородности поверхности. Важным фактором является то, что время соприкосновения частиц на катализаторе значительно больше, чем в газовой фазе, что также способствует ускорению протекания реакции.
Ферментативные реакции обычно относят к гетерогенно-каталитическим реакциям, в которых роль катализаторов выполняют ферменты. Ферменты состоят либо целиком, либо в основном из белков, то есть являются полимерами, образованными из аминокислот и имеющими определенную пространственную структуру из полипептидных цепей.
Ферменты обладают высокой специфичностью. Каждый фермент катализирует только определенный химический процесс или определенную группу превращений. Реакцию можно сравнить с замком, а фермент с ключом, и как не всяким ключом можно открыть любой замок, так и для ускорения данной реакции необходим свой фермент. Так, ферменты группы карбогидраз катализируют реакции гидролиза углеводов и практически не влияют на расщепление белков, в то время как фермент пепсин способствует расщеплению белков, но не влияет на протекание гидролиза углеводов.
Ферменты катализируют как процессы разложения, так и процессы синтеза, в частности синтеза белков. Каталитическая активность ферментов чрезвычайно высока. Так, время полупревращения для реакции разложения мочевины водой
СО(NH2)2 + H2O → CO2 + 2NH3.
при 25° С без катализатора составляет 109 с, а в присутствии фермента уреазы оно уменьшается до 10–4 с. Каталитическая активность ферментов намного выше, чем действие известных неорганических катализаторов. Например, при
25 оС под действием 1 моль фермента алкогольдигидрогеназы за одну секунду происходит окисление 720 моль спирта до уксусного альдегида, в то время как на промышленных катализаторах из меди за это время при 200 оС происходит окисление не более, чем одного моль спирта на 1 моль катализатора.
Каталитической активностью обладает не вся молекула фермента, а лишь определенный ее участок, называемый активным центром. Активный центр соединяется с молекулой реагирующего вещества, образуя непрочное промежуточное соединение, способное к дальнейшим превращениям. При этом активный центр вступает в соединение только с теми молекулами, структура которых подобна структуре активного центра. В состав небелковой части фермента могут входить ионы металлов и некоторые органические вещества. Если последние проявляют каталитическую активность, входя в активный центр фермента, то их называют коферментами.
Активность ферментов зависит от рН среды, концентрации фермента и температуры. Каждый фермент проявляет максимум активности при определенном значении рН. Повышение температуры приводит к увеличению активности фермента, однако при 40-50 °С достигается ее максимум. При дальнейшем увеличении температуры каталитическое действие ферментов ослабляется, так как начинается их тепловая денатурация.
Активность ферментов может быть снижена в результате воздействия ингибиторов. Существуют два типа ингибиторов – обратимые и необратимые. При обратимом ингибировании устанавливается равновесие между ферментом и ингибитором. При необратимом ингибировании активность фермента постепенно подавляется и может быть достигнута полная его инактивация, если концентрация необратимого ингибитора превышает концентрацию фермента.
Для описания кинетики ферментативных реакций используют уравнение, полученное Л. Михаэлисом и М. Ментен в 1913 г. Рассмотрим вывод данного кинетического уравнения для гомогенного каталитического процесса на примере реакции с одним исходным веществом (субстратом) S , которое превращается в продукт P в присутствии катализатора E (в частности, таким катализатором может быть фермент):

Через некоторе время от начала реакции, можно считать, что концентрация промежуточного комплекcа ES становится постоянной, то есть в системе наступает стационарное состояние. В этих условиях справедливо уравнение

где KM – константа Михаэлиса. Так как cE+сES=сo,E, где co,E – начальная концентрация фермента, то cE=сo,E-сES
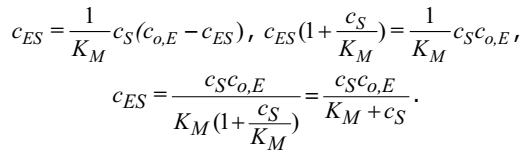
Скорость образования продукта реакции будет определяться выражением 
Согласно полученному уравнению, скорость реакциии прямо пропорциональна количеству катализатора и концентрации субстрата. Если cS<<KM, скорость образования продукта прямо пропорциональна cS . Если же cS>>KM, то скорость образования продукта реакции не зависит от cS и имеет максимальную величину. В этих условиях весь катализатор входит к состав промежуточного соединения ES , концентрация которого равна co,E, . Поэтому можно ввести величину максимальной скорости
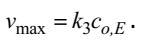

Это уравнение называют уравнением Михаэлиса–Ментен. Согласно этому уравнению, константа Михаэлиса численно равна концентрации субстрата, при которой скорость составляет половину от максимальной. Зависимость скорости каталитической реакции от концентрации субстрата при выполнения уравнения Михаэлиса– Ментен имеет вид, показанный на рисунке 1.4.
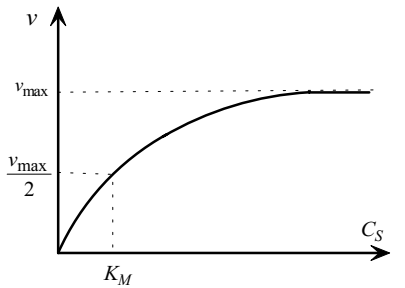
Рисунок 1.4 - Зависимость скорости каталитической реакции от концентрации субстрата
Величину vmax можно определить можно экспериментально, если удается провести кинетические измерения при условии cS>>KM. Скорость реакции при этом не зависит от концентрации субстрата до тех пор, пока последняя не уменьшится до уровня, соизмеримого с величиной KM.
Для определения vmax и KM удобнее использовать уравнение Михаэлиса–Ментен в следующем виде
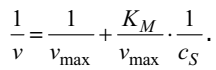
Эта форма уравнения получена Лайнуивером и Берком. Очевидно, что в координатах 1/v – 1/S c график этой зависимости будет линейным. Тангенс угла наклона этой прямой равен KM/vmax , а отрезок, отсекаемый на оси ординат – 1/vmax . Величина константы Михаэлиса KM для различных систем изменяется от 1 до 10–8 моль/л. Помимо природы субстрата, она зависит от величины рН, температуры и других факторов. Поэтому значение KM приводят для характеристики конкретных фермент–субстратных систем в определенных условиях. Ферментативный катализ играет огромную роль везде, где речь идет о живых организмах. Для повышения жизнедеятельности организма и улучшения обмена веществ создано много ферментативных препаратов, используемых в качестве лекарственных средств. Широкое применение получили ферментативные препараты при нарушениях функции желудочно-кишечного тракта, связанных с недостаточной выработкой пищеварительных ферментов. Так, при некоторых формах гастрита применяются препараты пепсин или панкреатин. Успешно применяются ферменты и в тех случаях, когда необходимо разрушить накопившиеся в большом количестве белковые образования (при ожогах, гнойных ранах, гнойно-воспалительных заболеваниях легких и т.д.). В этих случаях применяются протолитические ферменты, приводящие к быстрому гидролизу белков и способствующие рассасыванию гнойных скоплений. Для лечения ряда инфекционных заболеваний используются препараты лизоцима, которые разрушают оболочку некоторых болезнетворных бактерий. Очень важны ферменты, которые рассасывают тромбы (сгустки крови внутри кровеносных сосудов). Это плазмин, содержащийся в крови; ферменты поджелудочной железы – трипсин и химотрипсин. На их основе с разными добавками созданы лекарственные ферментные препараты – стрептокиназа, стрептаза и другие, широко применяемые в медицине.
Фотосинтез – это процесс трансформации поглощенной организмом энергии света в химическую энергию органических (и неорганических) соединений.
Процесс фотосинтеза выражают суммарным уравнением:
6СО2 + 6Н2О ® С6Н12О6 + 6О2,
в котором выражена суть явления, состоящая в том, что на свету в зеленом растении из предельноокисленных веществ - диоксида углерода и воды образуются органические вещества, и высвобождается молекулярный кислород. В процессе фотосинтеза восстанавливаются не только СО2, но и нитраты или сульфаты, а энергия может быть направлена на различные эндэргонические процессы, в том числе на транспорт веществ.
Общее уравнение фотосинтеза может быть представлено в виде:
12 Н2О → 12 [Н2] + 6 О2 (световая реакция)
6 СО2 + 12 [Н2] → С6Н12О6 + 6 Н2О(темновая реакция)
6 СО2 + 12 Н2О → С6Н12О6 + 6 Н2О + 6 О2
или в расчете на 1 моль СО2:
СО2 +Н2О  СН2О + О2
СН2О + О2
Весь кислород, выделяемый при фотосинтезе, происходит из воды. Вода в правой части уравнения не подлежит сокращению, так как ее кислород происходит из СО2. Методами меченых атомов было получено, что Н2О в хлоропластах неоднородна и состоит из воды, поступающей из внешней среды и воды, образовавшейся в процессе фотосинтеза. В процессе фотосинтеза используются оба типа воды. Доказательством происхождения О2 в процессе фотосинтеза служат работы голландского микробиолога Ван Ниля, который изучал бактериальный фотосинтез, и пришел к выводу, что первичная фотохимическая реакция фотосинтеза состоит в диссоциации Н2О, а не разложении СО2. Способные к фотосинтетической ассимиляции СО2 бактерии (кроме цианобактерий) используют в качестве восстановителей Н2S, Н2, СН3 и другие, и не выделяют О2. Такой тип фотосинтеза называется фоторедукцией:
СО2 + Н2S → [СН2О] + Н2О + S2 или
СО2 + Н2А → [СН2О] + Н2О + 2А,
где Н2А – окисляет субстрат, донор водорода (у высших растений – это Н2О), а 2А – это О2. Тогда первичным фотохимическим актом в фотосинтезе растений должно быть разложение воды на окислитель [ОН] и восстановитель [Н].При это [Н] восстанавливает СО2, а [ОН] участвует в реакциях освобождения О2 и образования Н2О. ]
Учение К.А. Тимирязева о космической роли зеленых растений. Фотосинтез, возникнув на первых этапах эволюции жизни, остается важнейшим процессом биосферы. Именно зеленые растения посредством фотосинтеза обеспечивают космическую связь жизни на Земле с Вселенной и определяют экологическое благополучие биосферы вплоть до возможности существования человеческой цивилизации. Фотосинтез - это не только источник пищевых ресурсов и полезных ископаемых, но и фактор сбалансированности биосферных процессов на Земле, включая постоянство содержания кислорода и диоксида углерода в атмосфере, состояние озонового экрана, содержание гумуса в почве, парниковый эффект и т. д.
Глобальная чистая продуктивность фотосинтеза составляет 78-108т углерода в год, из которых 7% непосредственно используют на питание, топливо и строительные материалы. В настоящее время потребление ископаемого топлива приблизительно сравнялось с образованием биомассы на планете. Ежегодно в ходе фотосинтеза в атмосферу поступает 70-120 млрд. т кислорода, обеспечивающего дыхание всех организмов. Одним из важнейших последствий выделения кислорода является образование озонового экрана в верхних слоях атмосферы на высоте 25 км. Озон (О3) образуется в результате фотодиссоциации молекул О2 под действием солнечной радиации и задерживает большую часть ультрафиолетовых лучей, губительно действующих на все живое.
Существенным фактором фотосинтеза является также стабилизация содержания СО2 в атмосфере. В настоящее время содержание СО2 составляет 0,03 % по объему воздуха, или 711 млрд. т в пересчете на углерод. Дыхание организмов, Мировой океан, в водах которого растворено в 60 раз больше СО2, чем находится в атмосфере, производственная деятельность людей, с одной стороны, фотосинтез - с другой, поддерживают относительно постоянный уровень СО2 в атмосфере. Диоксид углерода в атмосфере, а также вода поглощают инфракрасные лучи и сохраняют значительное количество теплоты на Земле, обеспечивая необходимые условия жизнедеятельности.
Однако за последние десятилетия из-за возрастающего сжигания человеком ископаемого топлива, вырубки лесов и разложения гумуса сложилась ситуация, когда технический прогресс сделал баланс атмосферных явлений отрицательным. Положение усугубляется и демографическими проблемами: каждые сутки на Земле рождается 200 тыс. человек, которых нужно обеспечить жизненными ресурсами. Эти обстоятельства ставят изучение фотосинтеза во всех его проявлениях, от молекулярной организации процесса до биосферных явлений, в ранг ведущих проблем современного естествознания. Важнейшие задачи - повышение фотосинтетической продуктивности сельскохозяйственных посевов и насаждений, а также создание эффективных биотехнологий фототрофных синтезов.
К.А. Тимирязев первым начал изучать космическую рользеленых растений. Фотосинтез – это единственный процесс на Земле, идущий в грандиозных масштабах и связанный с превращением энергии солнечного света в энергию химических соединений. Эта космическая энергия, запасенная зелеными растениями, составляет основу жизнедеятельности всех других гетеротрофных организмов на Земле от бактерий до человека. Выделяют 5 основных аспектов космической и планетарной деятельности зеленых растений.
1. Накопление органической массы. В процессе фотосинтеза наземные растения образуют 100-172 млрд.т. биомассы в год (в пересчете на сухое вещество), а растения морей и океанов – 60-70 млрд.т. Общая масса растений на Земле в настоящее время составляет 2402,7 млрд.т., причем 90% этой массы приходится на целлюлозу. Около 2402,5 млрд.т. приходится на долю наземных растений и 0,2 млрд.т. – на растения гидросферы (недостаток света!). Общая масса животных и микроорганизмов на Земле – 23 млрд.т., то есть 1% от массы растений. Из этого количества ~ 20 млрд.т. приходится на обитателей суши и ~ 3 млрд.т. – на обитателей гидросферы. За время существования жизни на Земле органические остатки растений и животных накапливались и модифицировались (подстилка, гумус, торф, а в литосфере – каменный уголь; в морях и океанах – толща осадочных пород). При опускании в более глубокие области литосферы из этих остатков под действием микроорганизмов, повышенных t0 и давления образовывались газ и нефть. Масса органических веществ подстилки ~ 194 млрд.т.; торфа – 220 млрд.т.; гумуса ~ 2500 млрд.т. Нефть и газ – 10000 – 12000 млрд.т. Содержание органического вещества в осадочных породах по углероду ~ 2 · 1016 т. Особенно интенсивное накопление органики происходило в палеозое (~ 300 млн. лет назад). Запасенное органическое вещество интенсивно используется человеком (древесина, полезные ископаемые).
2.Обеспечение постоянства содержания СО2 в атмосфере. Образование гумуса, осадочных пород, горючих полезных ископаемых выводили значительные количества СО2 из круговорота углерода. В атмосфере Земли становилось все меньше СО2 и в настоящее время его содержание составляет ~ 0,03% по объему или ~ 711 млрд.т. в пересчете на углерод. В кайнозое содержание СО2 в атмосфере стабилизировалось и испытывало лишь суточные, сезонные и геохимические колебания (стабилизация растений на уровне современных). Стабилизация содержания СО2 в атмосфере достигается сбалансированным связыванием и освобождением СО2 в глобальном масштабе. Связывание СО2 в фотосинтезе и образование карбонатов (осадочные породы) компенсируется выделением СО2 за счет других процессов: Ежегодное поступление СО2 в атмосферу (в пересчете на углерод) обусловлено: дыханием растений - ~ 10 млрд. т.: дыханием и брожением микроорганизмов - ~ 25 млрд.т.; дыханием человека и животных - ~ 1,6 млрд.т. хозяйственной деятельностью людей ~ 5 млрд.т.; геохимическими процессами ~ 0,05 млрд.т. Итого ~ 41,65 млрд.т. Если бы не происходило поступления СО2 в атмосферу, весь его наличный запас был бы связан за 6-7 лет Мощным резервом СО2 является Мировой океан, в его водах растворено в 60 раз больше СО2, чем его находится в атмосфере. Итак, фотосинтез, дыхание и карбонатная система океана поддерживает относительно постоянный уровень СО2 в атмосфере. За счет хозяйственной деятельности человека (сжигание горючих полезных ископаемых, вырубка лесов, разложение гумуса) содержание СО2 в атмосфере начало увеличиваться ~ на 0,23 % в год. Это обстоятельство может иметь глобальные последствия, так как содержание СО2 в атмосфере влияет на тепловой режим планеты.
3. Парниковый эффект. Поверхность Земли получает теплоту главным образом от Солнца. Часть этой теплоты возвращается в виде ИК лучей. СО2 и Н2О, содержащиеся в атмосфере, поглощают ИК лучи и таким образом сохраняют значительное количество теплоты на Земле (парниковый эффект). Микроорганизмы и растения в процессе дыхания или брожения поставляют ~ 85 % общего количества СО2, поступающего ежегодно в атмосферу и вследствие этого влияют на тепловой режим планеты. Тенденция повышения содержания СО2 в атмосфере может привести к увеличению средней t0 на поверхности Земли  таяние ледников (горы и полярные льды) затопление прибрежных зон. Тем не менее, возможно, что повышение концентрации СО2 в атмосфере будет способствовать усилению фотосинтеза растений, что приведет к связыванию избыточных количеств СО2.
таяние ледников (горы и полярные льды) затопление прибрежных зон. Тем не менее, возможно, что повышение концентрации СО2 в атмосфере будет способствовать усилению фотосинтеза растений, что приведет к связыванию избыточных количеств СО2.
4. Накопление О2 в атмосфере. Первоначально О2 присутствовал в атмосфере Земли в следовых количествах. В настоящее время он составляет ~ 21 % по объему воздуха. Появление и накопление О2 в атмосфере связано с жизнедеятельностью зеленых растений. Ежегодно в атмосферу поступает ~ 70-120 млрд.т. О2, образованного в фотосинтезе. Особую роль в этом играют леса: 1 га леса за 1 час дает О2, достаточно для дыхания 200 человек.
5. Образование озонового экрана на высоте ~ 25 км. О3 образуется при диссоциации О2 под действием солнечной радиации. Слой О3 задерживает большую часть УФ (240-290 нм), губительного для живого. Разрушение озонового экрана планеты – одна из глобальных проблем современности.
Структурная организация фотосинтетического аппарата
Лист как орган фотосинтеза . В процессе эволюции растений сформировался специализированный орган фотосинтеза - лист. Приспособление его к фотсинтезу шло в двух направлениях: возможно более полное поглощение и запасание лучистой энергии, и эффективный газообмен с атмосферой.
Рассмотрим, как складывается энергетический баланс листа в умеренной зоне. В летний полдень приход солнечной радиации составляет около 30-105 Дж/(м2.ч). В среднем листья поглощают 80-85 % энергии фотосинтетически активной радиации (ФАР), которой является видимая часть спектра электромагнитного излучения с длиной волны 400 -700 нм, и 25 % энергии инфракрасных лучей, что составляет около 55 % энергии общей радиации, или 16,5-105 Дж/(м2.ч). Лист отражает 10 % ФАР и 45 % инфракрасных лучей и пропускает соответственно 5 и 30 %.
На фотосинтез используется 1,5-2 (до 3) % поглощенной фотосинтетически активной радиации, остальная поглощенная энергия расходуется в основном на испарение воды - транспирацию (95- 98 %), возможен также и теплообмен с атмосферой.
Лист имеет ограниченный рост и характерное для данного вида и сорта строение. Как орган, осуществляющий ассимиляцию и испарение, он отличается плоской структурой и небольшой толщиной, измеряемой долями миллиметра. Благодаря этому при малых затратах строительного материала создается значительная общая поглощающая поверхность листьев. Так, сухая масса 1 м2 листовых пластинок составляет 30-40 г. Тонкая листовая пластинка лучше просвечивается, что способствует полноценной работе всех клеток листа.
Толщина листа тесно коррелирует с интенсивностью света, при которой он развивается. При ограниченном освещении толщина листовой пластинки меньше. При детальном рассмотрении поверхность листа выглядит волнистой, что увеличивает полноту улавливания солнечных лучей.
Листовая поверхность достигает значительных размеров и превосходит площадь почвы, которую занимает растение. Для характеристики размеров фотосинтетического аппарата используют индекс листовой поверхности, который рассчитывают как площадь листьев (м2), приходящуюся на 1 м2 почвы. Для сельскохозяйственных растений умеренной зоны средние значения листового индекса 3-5, в южных широтах с влажным климатом до 8-10. Например, поверхность листьев 1 га посевов зерновых культур в фазе цветения достигает 20-40 тыс. м2, у низкорослых яблонь во время вегетации площадь листьев составляет 25-30 тыс. м2 га.
Благодаря большой поверхности и определенному размещению листьев в пространстве растение может использовать как прямой, так и рассеянный свет, падающий под различными углами. Большое значение для эффективного улавливания света имеет архитектоника растений, под которой понимают пространственное расположение органов. У высокопродуктивных зерновых культур листья на стебле снизу вверх располагаются под все уменьшающимся углом и не затеняют друг друга. Оптимизация листовой поверхности посева или насаждения - важный способ управления продукционным процессом.
В зависимости от вида растений и условий их произрастания листья отличаются большим разнообразием. Однако можно выделить общие анатомические особенности, обеспечивающие возможность эффективного фотосинтеза.
1. Наличие покровной ткани - эпидермиса, защищающего лист от излишней потери воды. Клетки нижнего и верхнего эпидермиса лишены хлоропластов, имеют крупные вакуоли, которые, подобно линзам, фокусируют свет на расположенную глубже хлорофиллоносную ткань. Деятельность клеток мезофилла основана не только на их хорошем освещении, она зависит также от поступления СО2. Нижний эпидермис, реже и верхний, имеют большое количество устьиц. Щели открытых устьиц занимают примерно 1 % площади листовой пластинки, диффузия СО2 внутрь листа идет через них сравнительно быстро. Отдельное устьице позволяет за 1 с поступить в лист 2500 млрд. молекул СО2. Поверхность листа поглощает СО2 только в 1,5-2 раза меньше, чем открытая поверхность щелей той же площади, хотя открытые устьица составляют лишь сотую часть поверхности. Такая высокая скорость связана с особенностями диффузии газов через мелкие отверстия, находящиеся на значительном расстоянии друг от друга, за счет краевого эффекта.
2. Наличие специализированной фотосинтетической ткани - хлоренхимы. Основная хлорофиллоносная ткань - палисадная паренхима - расположена обычно на освещаемой части листа. Вытянутость клеток и перпендикулярное расположение их к эпидермису обеспечивают увеличение поверхности, вдоль которой могут располагаться хлоропласты, не затеняя друг друга, а также облегчают отток ассимилятов. В каждой клетке полисадной паренхимы находится 30-40 хлоропластов. Губчатая ткань характеризуется меньшим содержанием хлоропластов (примерно 20 на клетку) и сильно развитой системой межклетников. Объем межклетников составляет 15-20 % общего объема листа и образует внутреннюю газовую среду, которая при помощи устьичных щелей сообщается с атмосферой. За счет межклетников значительно возрастает внутренняя рабочая поверхность, через которую каждой клеткой паренхимы поглощается СО2. Она в 8-12 раз больше, чем наружная поверхность листа. Хлоропласты как основные светоулавливающие органеллы еще в большей степени увеличивают светопоглощающую поверхность листа. На 1 см2 листа приходится примерно 200 см2 поверхности хлоропластов. Это является выражением общебиологической особенности организации - создание больших внутренних рабочих поверхностей при сравнительно малых наружных испаряющих площадях за счет, как было отмечено, затрат небольших количеств материала.
3. Наличие сильно развитой густой системы жилок - проводящих путей, что обеспечивает быстрый отток ассимилятов и снабжение фотосинтезирующих клеток водой и необходимыми минеральными веществами.
В зависимости от внешних условий, при которых происходят формирование и функционирование листьев, анатомическое строение их может существенно различаться. Листья, формирующиеся в условиях недостаточной влагообеспеченности, имеют ксероморфную структуру. В зависимости от освещения меняется соотношение между полисадной и губчатой паренхимой в мезофилле. Имеются и другие приспособления для функционирования листа в определенных условиях. Еще более существенные отклонения от типичного строения листа связаны с физиолого-биохимическими особенностями фиксации СО2 у С4-растений, к которым относятся кукуруза, сахарный тростник, ряд злостных сорняков наших полей.
Основные элементы структуры хлоропластов . Лист растения - орган, обеспечивающий условия для протекания фотосинтетического процесса. Функционально же фотосинтез приурочен к специализированным органеллам - хлоропластам. Хлоропласты высших растений имеют форму двояковыпуклой линзы (диска), которая наиболее удобна для поглощения солнечных лучей. Их размеры, количество, расположение в клетке также полностью отвечают назначению: как можно эффективнее поглощать солнечную энергию, как можно полнее усваивать углерод. На различных растениях установлено, что количество хлоропластов в клетке измеряется десятками. Это обеспечивает высокое содержание этих органелл на единицу поверхности листа. Так, на 1 мм2 листа фасоли приходится 283 тыс. хлоропластов, у подсолнечника - 465 тыс. Диаметр хлоропластов в среднем 0,5-2 мкм, длина 5-10 мкм, объем 30- 40 мкм3. Малый размер хлоропластов и большое количество их в одной клетке обусловливают громадную общую суммарную рабочую поверхность.
Хлоропласты способны к активным движениям - изменению ориентации тела и перемещению в пространстве. Скорость движения хлоропластов около 0,12 мкм/с. Их передвижения вызываются физическими и химическими факторами. Например, под влиянием яркого света хлоропласты поворачиваются узкой стороной диска к падающим лучам и перемещаются на боковые стенки клеток. Хлоропластам присуща также хемотаксическая чувствительность - они передвигаются в направлении более высокой концентрации СО2 в клетке. Установлен и эндогенный суточный ритм движения хлоропластов: днем они обычно выстраиваются вдоль стенок, ночью опускаются на дно клетки.
Химический состав хлоропластов довольно сложен и характеризуется высоким содержанием воды (75 %). Около 75-80 % общего количества сухих веществ приходится на долю различных органических соединений, 20-25 % - на долю минеральных веществ. Структурной основой хлоропластов являются белки, содержание которых достигает 50-55 % сухой массы, примерно половину из них составляют водорастворимые белки. Такое высокое содержание белков объясняется их многообразными функциями в составе хлоропластов. Это структурные белки, являющиеся основой мембран, ферменты, транспортные белки, поддерживающие определенный ионный состав, отличающийся от цитозоля, сократительные белки, подобные актомиозину мышц, которые обеспечивают двигательную активность хлоропластов. Белки выполняют также рецепторную функцию, принимая участие в регуляции интенсивности фотосинтеза в меняющихся условиях внутренней и внешней среды.
Важнейшей составной частью хлоропластов являются липиды, содержание которых колеблется от 30 до 40 % сухой массы. Липиды хлоропластов представлены тремя группами соединений.
1. Структурные компоненты мембран, которые представлены амфипатичными липоидами и отличаются высоким содержанием (более 50 %) галактолипидов и сульфолипидов. Фосфолипидный состав характеризуется отсутствием фосфатидилэтаноламина и высоким содержанием фосфатидилглицерина (более 20 %). Свыше 60 % состава жирных кислот приходится на линолевую кислоту.
2. Фотосинтетические пигменты. В отличие от водорастворимых пигментов клеточного сока пигменты хлоропластов характеризуются повышенным сродством к гидрофобным растворителям и относятся к липоидам. Высшие растения содержат две формы зеленых пигментов: хлорофилл а и хлорофилл b и две формы желтых пигментов: каротины и ксантофиллы (каротиноиды). Ведущую роль в фотосинтезе - фотосенсибилизацию - выполняет хлорофилл а, другие пигменты расширяют спектр действия фотосинтеза за счет более полного поглощения ФАР, каротиноиды защищают хлорофилл от фотоокисления, участвуют в транспорте кислорода, образующегося при фотолизе воды.
3. Жирорастворимые витамины - эргостерол (провитамин D ), витамины Е, К сосредоточены практически целиком в хлоропластах, где участвуют в преобразовании световой энергии в химическую.
Нуклеиновые кислоты составляют примерно 1% сухой массы хлоропластов (РНК - 0,75 %, ДНК - 0,01-0,02%). Таким образом, хлоропласты имеют собственную белоксинтезирующую систему. Его геном представлен кольцевой молекулой ДНК длиной 40 мкм с молекулярной массой 108, кодирующей 100-150 белков средних размеров. Рибосомы хлоропластов составляют от 20 до 50% общей популяции рибосом клетки.
Углеводы не являются конституционными веществами хлоропласта. В очень небольших количествах фосфорные эфиры сахаров участвуют в восстановительном цикле углерода, в основном же это продукты фотосинтеза. Поэтому содержание углеводов в хлоропластах значительно колеблется (от 5 до 50 %). В активно функционирующих хлоропластах углеводы обычно не накапливаются, происходит их быстрый отток. При уменьшении потребности в продуктах фотосинтеза в хлоропластах образуются крупные крахмальные зерна. В этом случае содержание крахмала может возрасти до 50% сухой массы и активность хлоропластов снизится.
Необходимо обратить внимание такжена высокое содержание в хлоропластах минеральных веществ. Сами хлоропласты составляют 25-30 % массы листа, но в них сосредоточено до 80 % железа, 70-72 - магния и цинка, около 50 - меди, 60 % кальция, содержащихся в тканях листа. Эти данные хорошо согласуются с высокой и разнообразной ферментативной активностью хлоропластов. Минеральные элементы выступают в роли простетических групп и кофакторов деятельности ферментов. Магний входит в состав хлорофилла. Важная роль кальция состоит в стабилизации мембранных структур хлоропластов.
.При выращивании сельскохозяйственных растений следует иметь в виду, что на структуру хлоропластов, а, следовательно, и их функциональную активность большое влияние оказывает режим минерального питания растений. При недостатке азота хлоропласты становятся в 1,5-2 раза мельче, дефицит фосфора и серы нарушает нормальную структуру ламелл и гран, одновременная нехватка азота и калия приводит к переполнению хлоропластов крахмалом из-за нарушения нормального оттока ассимилятов. При недостатке кальция нарушается структура наружной мембраны хлоропласта. Для поддержания структуры хлоропласта также необходим свет. В темноте идет постепенное разрушение тилакоидов гран и стромы. Таким образом, структура хлоропластов лабильна и динамична, в ней отражаются все условия жизни растения.
Фотосинтез связан с избирательным поглощением пигментами света в видимой части солнечного спектра. Фотосинтетические пигменты составляют 10-15 % сухой массы хлоропластов. Они характеризуются большим разнообразием и по химической природе делятся на две группы - хлорофиллы и каротиноиды. В настоящее время известно несколько различных форм хлорофилла, которые обозначают латинскими буквами. Хлоропласты высших растений содержат хлорофилл а и хлорофилл b . Они были идентифицированы русским ученым М.С. Цветом (1906) с помощью разработанного им метода хроматографии. Структурная формула хлорофилла, предложенная Фишером (1939), получила окончательное подтверждение в 1960г. в результате двух независимо проведенных работ в США и ФРГ по искусственному синтезу хлорофилла а.
Оптические свойства. Согласно закону Гротгуса - одному из фундаментальных законов светохимии фотохимически деятельны только поглощенные лучи. Поэтому изучение спектров поглощения пигментов, и в первую очередь хлорофилла, является решающим для понимания механизма фотосинтеза и разработки путей его регуляции. Отличительной особенностью всех пигментов является наличие в их составе системы слабоудерживаемых делокализованных электронов, возбуждаемых квантами видимой части солнечного спектра. Это лежит в основе их свойства - избирательного поглощения света. Резко выраженные максимумы поглощения хлорофиллов находятся в сине-фиолетовой и красной частях спектра. Хлорофиллы очень слабо поглощают оранжевые и желтые лучи и совсем не поглощают зеленые и инфракрасные. Поэтому раствор хлорофилла а имеет сине-зеленый цвет, хлорофилла b - желто-зеленый.
Механизмы избирательного поглощения света и флуоресценции изучены достаточно хорошо. Поглощение кванта света сопровождается переходом в более богатое энергией короткоживущее возбужденное состояние, связанное с переходом электрона на более удаленную от ядра орбиталь (хлорофилл → хлорофилл*). Электронные орбитали атомов характеризуются определенными энергетическими уровнями, возрастающими по мере удаления от ядра. Те кванты света, энергия которых соответствует разности энергий между двумя орбиталями, поглощаются с переходом электрона на более дальнюю орбиталь. Поэтому могут поглощаться только кванты света с совершенно определенной длиной волны.
В органических молекулах почти все электроны спарены, т.е. находятся в одном и том же энергетическом состоянии, обладая противоположными спинами (основное синглетное энергетическое состояние). Поглощение молекулой хлорофилла кванта красного света с энергией 170 кДж/моль квантов приводит к первому синглетному электронно-возбужденному состоянию – S1, время существования которого примерно 10-9с. Возбужденная молекула хлорофилла переходит в стабильное состояние путем возвращения электрона на исходную орбиталь. Триплетное состояние длится гораздо дольше (> 10-4с). Из триплетного состояния молекула может вернуться в основное за счет более длительного, чем флуоресценция, слабого длинноволнового свечения - фосфоресценции или направив энергию на фотохимические реакции.
Сущность световой фазы фотосинтеза состоит в поглощении лучистой энергии и ее трансформации в ассимиляционную силу (АТФ и НАДФ-Н), необходимую для восстановления углерода в темновых реакциях. Сложность процессов преобразования световой энергии в химическую требует их строгой мембранной организации. Световая фаза фотосинтеза происходит в гранах хлоропласта.
Продукты световой фазы фотосинтеза используются в темновой фазе для восстановления СО2 до уровня углеводов. Реакции восстановления происходят настолько быстро, что с помощью обычных методов химического анализа чрезвычайно трудно обнаружить промежуточные продукты и практически невозможно установить последовательность их превращений. Только использование радиоактивных изотопов, в первую очередь 14С, в качестве метки, хроматографии и электрофореза позволило установить последовательность реакций при фотосинтезе, множественность путей метаболизма углерода.
Дата добавления: 2020-11-23; просмотров: 201; Мы поможем в написании вашей работы! |

Мы поможем в написании ваших работ!